


1. Introduction
Lithium secondary batteries are widely used in power mobile devices; the range of their applications is being extended to include transportation systems such as electric vehicles. However, the increasing sophistication of the electronics involved in these devices and the corresponding high demands for longer battery life have triggered investigations aimed at identifying ways to increase the energy density of these batteries [1]. In particular, substantial efforts are focusing on developing materials with performance superior to graphite, the current main active component of lithium secondary battery anodes, which has a theoretical capacity of 372 mAh/g. Silicon, tin, and transition metal oxides are possible candidates to replace graphite [2-4].
Tungsten oxide is a transition metal compound with strong potential as high-capacity anode material. While the hexagonal tungsten trioxide (WO3) cathode, whose lithium insertion process involves an intercalation mechanism, has also attracted some interest [5], many polymorphic forms of WO3 (such as monoclinic [6], tetragonal [7], triclinic, and amorphous structures [8]) have been evaluated as high-performance anode materials in lithium batteries. In this case, the reaction of 1 mol of WO3 with 6 mol of lithium, according to the conversion mechanism described below, can provide a theoretical capacity of 693 mAh/g, 1.9 times greater than the theoretical capacity of graphite [6,7]:
WO3 + 6Li+ + 6e− → W + 3Li2O charge (lithiation)
W + 3Li2O → WO3 + 6Li+ + 6e− discharge (delithiation)
However, the practical applications of tungsten oxide are limited, due to its relatively low conductivity [9] and the low electrochemical reaction rate at the electrode-electrolyte interface [10], as well as to the capacity decrease caused by the mechanical damage associated to volume contraction/expansion during charge/discharge [8].
Electrochemical deposition is widely used for the preparation of functional thin films, because it does not require expensive equipment, and the morphology of the films can be controlled by adjusting synthesis conditions such as temperature, bath chemistry, and size/type of driving force (e.g., current vs. voltage, continuous vs. pulsed). Many studies have been devoted to the preparation of compact WO3 thin films using electrodeposition and to their application in electrochromic devices or gas sensor electrodes [11-14]. A recent study on the application of electrodeposited WO3 thin films as anodes in lithium secondary batteries reported that the capacity per surface area and the ability to retain capacity of these materials were relatively low [8]. This finding could be attributed to the large overpotential induced by the high values of the electrical conductive and interfacial charge transfer resistances of compact tungsten oxide thin films. In addition, the stress induced by the change in the thin film volume during the reaction with lithium might cause its detachment from the substrate.
Typical strategies adopted to address these issues include decreasing the electrical conductive resistance by thinning the film, reducing the interfacial charge transfer resistance by creating a nanoporous structure or small particles with large specific surface area, and introducing spaces or pores large enough to effectively accommodate changes in volume [15-20]. Some attempts have been made to create nanoporous tungsten trioxides using porous templates, and outstanding electrochemical properties have been reported [7]. However, the high cost of the templates prevents the large-scale production of nanoporous tungsten trioxide and introduces further issues, such as the need for a sophisticated production process involving application and removal of templates, and the inability to control the porosity.
The present work aims at producing nanoporous WO3 using template-free electrodeposition, and at evaluating its effectiveness as anode material in lithium secondary batteries. The plating conditions were modified to produce thin films with different porosities and particle sizes. The electrochemical properties of the samples were determined by constant-current charge/discharge tests, and the results were used to discuss morphological effects on the performance of tungsten trioxide anodes in lithium secondary batteries.
2. Experimental Section
2.1. Preparation of Electrodeposited Tungsten Oxide Thin Films
The electrolytes used for the synthesis of tungsten oxide thin films by electrodeposition were prepared by two different methods, described below:
(1) 3.6 g of tungsten powder (Alfa, 99.9 %) was completely dissolved in 120 mL of H2O2 (Junsei, 30 wt%) and residual H2O2 was removed by decomposing it using a Pt mesh immersed in the solution for 24 h. The solution was then combined with a mixture of isopropanol (IPA, Honeywell, 99.99 %) and distilled water in a 1:1 volume ratio to create a 50 mM tungsten oxide precursor solution [8,21]. This solution, with an IPA concentration of 20 vol.%, was then used to create a dense tungsten oxide thin film.
(2) Following a procedure similar to (1), 4.6 g of tungsten powder was dissolved in 25 mL of H2O2 and residual H2O2 was removed from the solution using a Pt mesh. Ethylene glycol (EG, Alfa, 99 %) and distilled water were then added to the solution to create a 50 mM tungsten oxide precursor, with an EG concentration of 50 or 75 vol.%. The electrolytes were kept at room temperature or at 50 ℃ [22]. These solutions were used for preparing porous tungsten oxide thin films.
A three-electrode cell with a saturated calomel electrode (SCE) as reference electrode was employed for the electrodeposition. A 304 stainless steel foil (Alfa Aesar, Fe/Cr/Ni = 70:19:11, wt.%) was used as the substrate (anode), and a Pt wire was used as the cathode. The surface of the substrate was treated with HCl solution (10 wt.%) before plating, to remove the native oxide layer, and the surface area was set to 2.5 × 10−5 m2. Pulse plating was used to produce a dense thin film, following previous indications that flaws (such as cracks) on the surface of a tungsten oxide thin film are greatly reduced when a pulsed voltage is applied [8]. The following conditions were employed: applied potential, −1.0 V vs. SCE; on-time duration (ton), 0.01 s; duty cycle (ton / (ton + toff) × 100, where toff is the offtime duration), 50%; number of pulse applications, 3000. A 200 nm-thick compact layer was produced under these conditions. Electrodeposited porous thin films were synthesized by applying a voltage of −0.6 V vs. SCE [22]. The thickness of the porous layers was set to 200-300 nm on average, similar to that of a dense thin film, by adjusting the voltage application time. An IviumStat system (Ivium Technologies, Netherlands) was used for the electrodeposition. Some of the porous thin films prepared were heat-treated at 450 ℃ for 6 h to improve their adhesion to the substrate.
Surface and cross-section morphologies of the synthesized tungsten oxide thin films were observed using a scanning electron microscope (MIRA3, TESCAN, Czech Republic). The porosity of the samples was determined by measuring the weight gain upon filling the pores with propylene carbonate (PC) under decompression at ~13 kPa [23,24]. The surface area and particle sizes were measured using a specific surface area analyzer (TriStar II 3020 surface area and porosity system, Micromeritics, USA). The tungsten oxide composition was analyzed using X-ray photoelectron spectroscopy (XPS) using an ESCALAB 250 spectrometer (VG Scientifics, USA), and the crystal structure was analyzed using an X-ray diffractometer (X'Pert PRO, PANalytical, Netherlands).
2.2. Electrochemical Tests
To determine the temperature dependence of the elementary resistances during the electrodeposition of tungsten oxide, electrochemical impedance spectroscopy (EIS) measurements at 25, 50, and 75 ℃ were performed at the open-circuit potential with potential fluctuations of 5 mVrms, in a frequency range of 0.01-106 Hz. To evaluate the performance of electrodeposited tungsten trioxide as anode material for lithium secondary batteries, a three-electrode beaker cell was assembled, with lithium reference and counter electrodes. The cell electrolyte was a 1 M solution of lithium hexafluorophosphate (LiPF6) in a 1:1 (v/v) mixture of ethylene carbonate and dimethyl carbonate. The initial capacity and the capacity retention of the WO3 thin films were analyzed through a constant-current charge/discharge test at a rate of 0.1 C (the C rate was calculated on the basis of the theoretical capacity of WO3). To estimate the interfacial resistances, the EIS measurements were conducted at the open circuit potential with a potential fluctuation of 5 mVrms over the frequency range of 0.01-106 Hz. The beaker cell assembly and the related electrochemical tests were carried out inside a glove box (MBraun, Germany) filled with high-purity Ar gas, and the constant-current charge/discharge tests were performed using a CTS-LAB battery test system (BaSyTec GmbH, Germany). A VMP3 system (BioLogic Co., Claix, France) was used for the EIS tests.
3. Results and Discussion
Fig. 1a shows the tungsten oxide thin film prepared in the room-temperature electrolyte containing 20 vol.% IPA. The film appears relatively smooth and uniform, with no noticeable flaws. Compared to this film, the thin film prepared in the electrolyte containing 50 vol.% EG shows a higher surface roughness (Fig. 1b). It has been reported that when the diffusion of active species towards the electrode-electrolyte interface is the rate-controlling step of the electrodeposition, an increased surface roughness of the plated layer may result, and the layer can even form a dendritic structure in extreme cases [25-27]. Since EG is eight times more viscous than IPA (the viscosities of IPA and EG are 1.96 and 16.1 cP, respectively), its use might increase the surface roughness of the synthesized plated layers by restricting the movement of the peroxytungstate ion (W2O112−) in the electrolyte and thereby limiting the overall electrodeposition rate. The different morphology of the thin film electrodeposited using EG is consistent with the results reported in the literature [22].
Fig. 1.
Surface and 30° inclined (insets) views of tungsten oxide films deposited at 25 ℃ in a solution containing (a) 20 vol.% isopropanol (IPA), (b) 50 vol.% ethylene glycol (EG), and (c) 75 vol.% EG. (d) Microscopic images of tungsten oxide films deposited at 50 ℃ in a solution containing 50 vol.% EG..

Assuming that a diffusion-controlled electroplating process causes the surface to change from smooth to rough, a more diffusion-controlled environment could represent a valid solution for obtaining the desired porosity by enhancing the surface roughness. This goal could be achieved through two possible experimental routes: (1) the diffusion rate of active ions could be further reduced by increasing the fraction of EG, and (2) the interfacial electrochemical reaction rate could be increased, compared to the ion diffusion rate, by adjusting the temperature (this method can be applied only when the difference in activation energy between the interfacial and ion diffusion processes is large). Figs. 1c and 1d show the morphologies of the plated layers when the EG content was raised from 50 % to 75 % and when the temperature was raised to 50 °C at a fixed EG concentration, respectively. Porous thin films were formed in both cases. Interestingly, increasing the temperature (Fig. 1d) appears more effective at developing the porosity of the plated layer than increasing the EG content (Fig. 1c). Therefore, under the conditions applied in the present experiments, it appears that increasing the temperature is more effective at producing a diffusion-controlled environment than increasing the EG content.
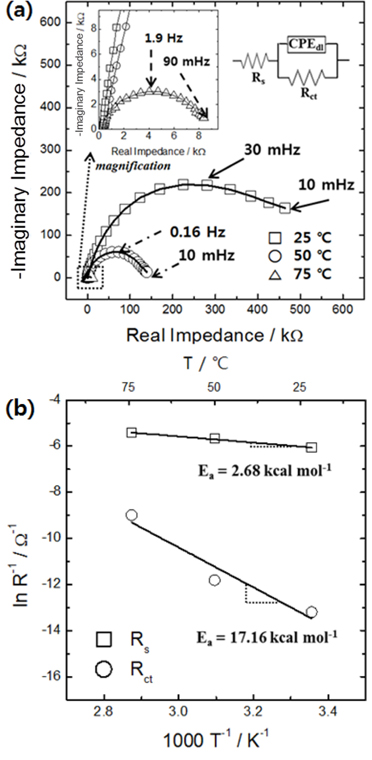
As a matter of fact, it has been reported that the porosity of tungsten oxide increases with increasing temperature. However, this effect has been rather vaguely attributed to “enhanced deposition kinetics” without any supporting experimental data [22]. Because “deposition kinetics” can encompass both interfacial charge transfer and ion diffusion, it is necessary to individually assess the rates and resistances associated to both processes in order to explain the effect of temperature in more detail. The formation of a more diffusion-controlled environment with increasing temperature indicates that the temperature sensitivity of the interfacial electrochemical reaction rate (in other words, its activation energy) is greater than that of the ion diffusion rate in the electrolyte. In order to confirm this suggestion, the temperature dependence of the elemental resistances during tungsten oxide electrodeposition was analyzed in detail using an impedance method (Fig. 2a). The electrolyte resistance decreased marginally with increasing temperature, whereas the charge transfer resistance at the interface decreased significantly. Arrhenius plots of these data (Fig. 2b) show that the activation energy for the interfacial charge transfer reaction was at least six times larger than that for ion transport in the electrolyte. The difference between the activation energies or between the temperature sensitivities of the elementary resistances highlights that increasing the temperature leads to a larger increase in the interfacial electrochemical reaction rate than in the ion transport rate during tungsten oxide deposition. The results also confirm that increasing the temperature is a very effective way to increase the porosity of tungsten oxides, by creating a more diffusion-controlled environment.
Fig. 2.
(a) Impedance spectra obtained at various temperatures during electrolytic deposition of tungsten oxide films; (b) Arrhenius plots of ohmic (Rs) and charge transfer (Rct) resistances. The inset in panel (a) shows the equivalent circuit used for analyzing the impedance spectra.
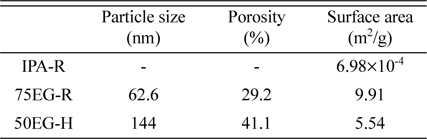
Among the four tungsten oxide films displayed in Fig. 1, we focused on the following three films, which exhibited relatively large morphological differences: the compact film prepared at room temperature in an IPA-containing electrolyte (Fig. 1a, IPA-R hereafter), the porous film created at room temperature in an electrolyte containing 75 vol.% EG (Fig. 1c, 75EG-R hereafter), and the porous film created at 50 ℃ in an electrolyte containing 50 vol.% EG (Fig. 1d, 50EG-H hereafter). Table 1 shows the morphological characteristics of these three films. The porosity of 50EG-H was about 10 % greater than that of 75EG-R, which is consistent with the qualitative results shown in Fig. 1. However, 50EG-H exhibited a larger particle size and lower surface area per weight than 75EG-R, which may be the result of the enhanced particle growth induced by the faster deposition reaction at higher temperature. This implies that 50EG-H might be partly more effective than 75EG-R as a lithium battery anode because its higher porosity and the corresponding higher amount of space available within the electrodes can allow it to maintain the integrity of the entire anode by accommodating volume expansion/contraction processes during the battery operation. On the other hand, the smaller particle size would make the 75EG-R film more effective as anode, because smaller particles can better withstand mechanical breakage when stress increases during the battery operation. Therefore, it is hard at this stage to predict how different the actual capacity retention of each film will be, owing to the contrasting effects of porosity and particle size.
Table 1.
Particle size, porosity, and surface area of tungsten oxides deposited in solutions containing 20 vol.% isopropanol (IPA) at 25 ℃ (IPA-R), 75 vol.% ethylene glycol (EG) at 25 ℃ (75EG-R), and 50 vol.% EG at 50 ℃ (50EG-H).
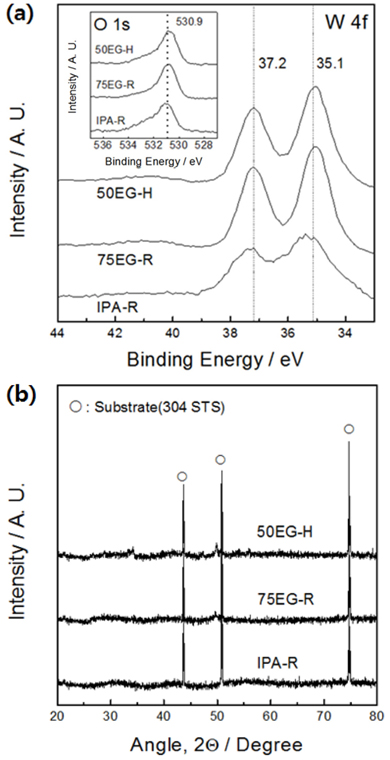
The results of the XPS analysis of the electrodeposited tungsten oxides are shown in Fig. 3a. For all specimens, the binding energies corresponding to W 4f7/2 and W 4f5/2 (35.1 and 37.2 eV, respectively) coincide with the binding energies of W6+ in WO3 [6]. In addition, the O 1s peak, corresponding to the metal oxide binding energy, lies exactly at the expected location (530.9 eV) (inset of the figure). These results confirm that all thin films synthesized in this work were composed of tungsten trioxide. The crystal structure analysis showed that no crystalline phases apart from the substrate were present (Fig. 3b), thus revealing that the synthesized thin films had an amorphous structure with only short-range order.
Fig. 3.
(a) W 4f and O 1s (inset) X-ray photoelectron spectroscopy (XPS) spectra of tungsten oxide films deposited in a solution containing 20 vol.% isopropanol (IPA) at 25 ℃ (IPA-R), 75 vol.% ethylene glycol (EG) at 25 ℃ (75EG-R), and 50 vol.% EG at 50 ℃ (50EG-H). (b) X-ray diffraction patterns of the samples.
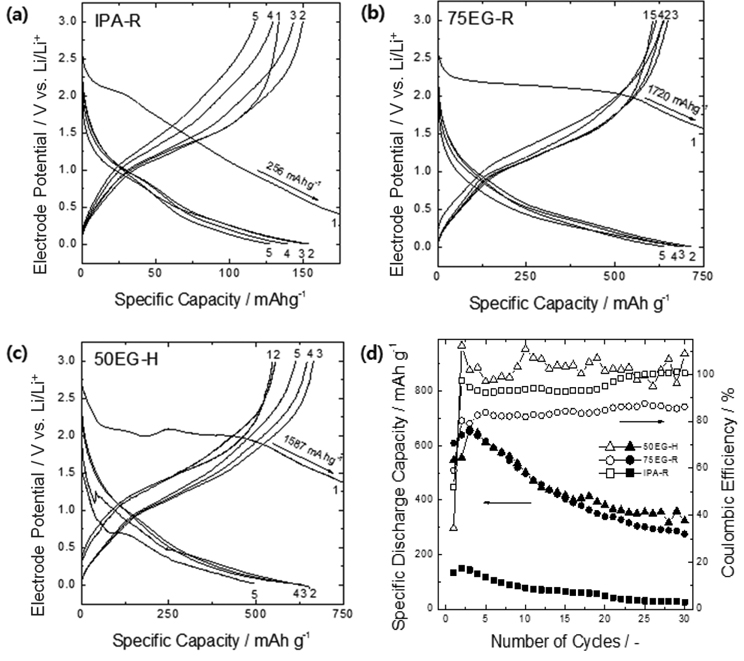
The three electrodeposited WO3 films with different morphologies (IPA-R, 75EG-R, 50EG-H) were evaluated as lithium secondary battery anodes. Figs. 4a-c present the initial five constant-current charge/discharge curves for the three samples. After the first charge (lithiation) process, where a large capacity loss probably due to irreversible surface reactions was observed, and the subsequent two or three discharge/charge (i.e., activation) processes, a reversible reaction was confirmed to take place between all three specimens and lithium. The initial discharge (delithiation) capacity of the dense IPA-R thin film remained around 150 mA h/g, whereas the capacities of the porous 75EG-R and 50EG-H thin films were much higher (above 600 mA h/g after the activation process). Considering that the amorphous structure doesn’t provide well-defined interstitial sites for lithium transport and the specific capacities of the porous films in this work were much higher than the capacity that is expected in typical intercalation compounds [5], the reaction of our electrodeposited films with lithium most likely proceeds according to the conversion mechanism [6-8].
Compared to compact oxides, porous oxides have low interfacial charge transfer resistance because of their large surface area, which in turn produces a low activation polarization. In addition, the short characteristic lithium diffusion length within the oxides decreases the diffusion resistance: in a 200 nm-thick compact flat-type oxide, the characteristic Li ion diffusion length within the oxide is 200 nm (without taking into account the change in volume), whereas the characteristic length within a spherical porous oxide of 63-144 nm diameter (Table 1) is 11-24 nm (estimated as particle radius/3). In summary, the porous thin films used in this work possibly have lower interfacial charge transfer and solid-phase diffusion resistances than the dense thin film, which in turn led to a lower overvoltage during the discharge (delithiation) process and thus a lower discharge potential. These features increase the film capacity by providing more opportunities for lithium alloying/dealloying during charge/discharge processes.
Fig. 4d shows the change in Coulombic efficiency and capacity retention with the number of charge/discharge cycles. The IPA-R film shows a dramatic decrease in capacity with increasing number of charges and discharges: its capacity was reduced to 26 mA h/g, 17 % of the initial capacity, after 30 cycles. In contrast, the porous 75EG-R and 50EG-H specimens showed much higher capacity retentions of 42 % (275 mA h/g) and 49 % (324 mA h/g), respectively, compared to the dense thin film. Considering that the capacity of 50EG-H slightly fluctuated in the final cycles, it seems reasonable to conclude that there was virtually no difference in capacity retention between 75EG-R and 50EG-H. This result reveals that, at least for the WO3 thin films examined in this work, when it comes to withstanding breakage during charge and discharge, the effect of a smaller particle size (as for the specimen 75EG-R) is comparable to the effect of a larger free volume available within the electrode (as for the specimen 50EG-H).
Fig. 4.
Typical voltage profiles of the as-deposited tungsten oxide anodes (a) IPA-R, (b) 75EG-R, and (c) 50EG-H for the first five cycles at a charge/discharge rate of 0.1 C. (d) Dependence of their specific discharge capacities and Coulombic efficiencies on cycle number.
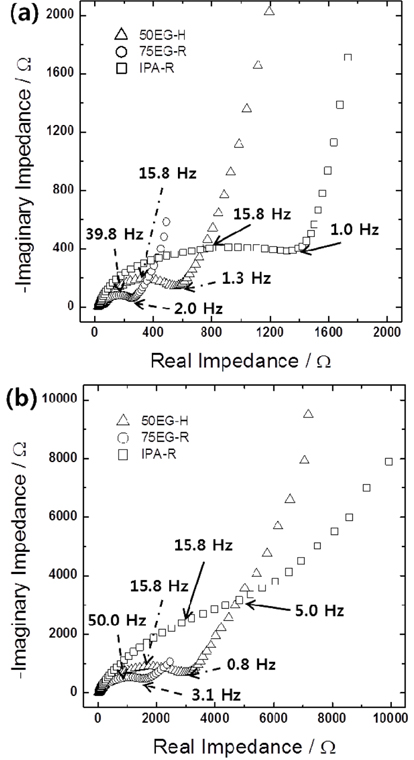
Figs. 5a and b show the Nyquist plots obtained for IPA-R, 75EG-R, and 50EG-H in lithiated and delithiated states, respectively. It was not possible to separate the resistances corresponding to lithium ion migration through the solid electrolyte interface and to interfacial charge transfer, because the arcs corresponding to the two processes overlapped in the semicircle from the high to intermediate frequency region. Nevertheless, the total interfacial resistances of three samples could be roughly compared from the size of the depressed semicircle. The interfacial resistances of 75EG-R and 50EG-H were much lower than the corresponding resistances of IPA-R. This result is most likely due to the much higher surface areas of the former porous films than the latter dense one. Two further points are worth noting. One is the lower interfacial resistance of 75EG-R than 50EG-H, regardless of their lithiated and delithiated states, which is definitely due to the larger surface area (or smaller particle size) of 75EG-R. The other is the significantly smaller resistances in lithiated than delithiated state. Considering that the impedance spectra of porous thin films in lithiated state were measured at around 0.1 V vs. Li/Li+ (see the figure caption for details), the availability of a large number of reaction sites for additional alloying/dealloying due to incomplete lithiation might be partly responsible for the smaller interfacial resistances in the lithiated state. In addition, the volume expansion of the individual particles and the resulting increase in surface area could lead to a further decrease in the interfacial resistances.
Fig. 5.
Impedance spectra of IPA-R, 75EG-R, and 50EG-H samples in (a) lithiated (charged) and (b) delithiated (discharged) states. The open circuit potentials during the measurements were 0.38, 0.12, 0.11 V vs. Li/Li+ and 2.65, 2.06, 2.32 V vs. Li/Li+ in lithiated and delithiated states, respectively.
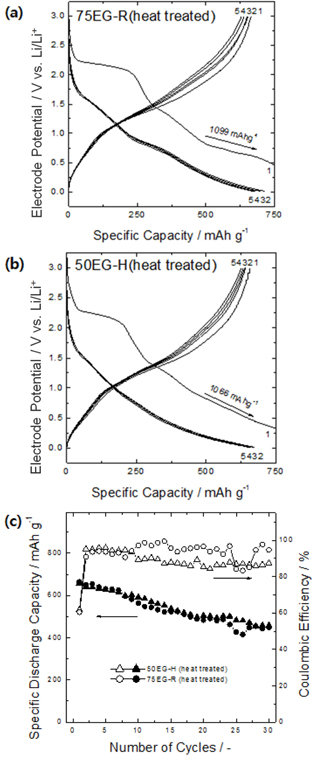
The performance of porous thin films that experience large volume changes during charge/discharge processes could be further enhanced by improving the adhesion between the film and the substrate. Thus, the present porous thin films were heat-treated to strengthen the film-substrate adhesion via atomic diffusion across the interface and thus suppress the detachment of the film from the substrate. Figs. 6a and b present the charge/discharge curves of the heat-treated porous oxides. The initial capacities of these samples were almost identical to those before heat treatment, but the reversibility of the reaction after the first charge was significantly improved. As shown in Fig. 6c, the capacity retention of the heat-treated samples after 30 cycles was greatly improved compared to that before heat treatment, and the capacity at the 30th cycle was about 1.2 times greater than the theoretical capacity of graphite (448 mA h/g and 458 mA h/g for 75EG-R and 50EG-H, respectively). Direct observation of the as-deposited porous thin film after the charge/discharge cycles revealed that the film became partially detached in several areas (Fig. 7a). On the other hand, the mechanical breakage and detachment of the plated layer after the charge/discharge cycles were greatly reduced after heat treatment (Fig. 7b).

4. Conclusions
This work aimed at producing porous tungsten trioxide thin films through electrodeposition and evaluating their properties as anodes in lithium secondary batteries. The following conclusions were drawn:
(1) Increasing the temperature is more effective in forming tungsten oxide films with high porosity than increasing the electrolyte viscosity. The analysis of the changes in interfacial charge transfer and electrolyte resistances with temperature revealed that the temperature sensitivity of the charge transfer reaction rate (i.e., of the activation energy) was much higher than that of the ion transport rate in the electrolyte. This finding implies that increasing the electrolyte temperature results in a more diffusion-controlled electrodeposition process, because the interfacial electrochemical reactions become much easier than the ion diffusion.
(2) The initial discharge capacities of the porous WO3 thin films were about four times greater than those of dense thin films. This was attributed to the reduced interfacial charge transfer and solid-phase diffusion resistances of porous thin films, due to their higher surface area and shorter characteristic diffusion length, respectively.
(3) The analysis of the capacity retention properties of the porous thin films revealed that, at least for the ranges of particle sizes and porosities used in this work, both high porosity and small particle size represent favorable factors leading to excellent capacity retention.
(4) The capacity retention of the porous thin films was greatly improved after heat treatment. Their reversible discharge capacity at the 30th cycle was more than 1.2 times greater than the theoretical capacity of graphite. Direct observation of the thin films after the charge/discharge cycles revealed that mechanical breakage and detachment of the film were greatly reduced after heat treatment, due most likely to the improved adhesion between the film and substrate.




