


1. Introduction
Molten carbonate fuel cells (MCFCs) have been consistently developed for decades owing to their high efficiency and low pollutant emission; currently, they are poised on the brink of commercialization. To ensure economic feasibility, a guarantee of 40,000 h of operation is required. The lifetime of an MCFC stack depends on the stabilities of its components including the cathode, anode, matrix, separator, and current collector in the carbonate melts [1], as well as the electrolyte loss caused by volatilization and corrosion in the metallic separators [2,3]. Many researchers have attempted to improve the stability of the components [4-6] and minimize electrolyte loss using various methods [7]. One of the simplest methods for minimizing electrolyte loss and increasing the span of an MCFC stack is to operate it at lower temperatures. Comparing the results after 33,000 h of operation at 650°C with those after 66,000 h of operation at 600°C, Morita et al. [7] demonstrated that MCFC can achieve a lifetime of over 40,000 h when operated at lower temperatures.
However, operation at lower temperatures impairs the stack performance, mainly because of slower reaction kinetics. The oxygen reduction reaction (ORR) on the cathode is considerably slower than the hydrogen oxidation reaction on the anode; therefore, the stack performance depends primarily on the cathode reaction [8,9]. Thus, it is necessary to enhance the electrochemical catalytic activity of the cathode for long-term operation at low temperatures without degrading the performance of the MCFC system.
In MCFC cathodes, the ORR occurs via the following steps [10-12]: (1) oxygen in the gas phase dissolves in the carbonate melts in the form of superoxide (O2−) or peroxide (O22−) ions; (2) these ions are reduced to oxygen ions (O2−) by removal of electrons from the electrode; and (3) the oxygen ions react with CO2 to form carbonate ions (CO32−). Of these, the second step, i.e., the formation of oxygen ions from superoxide or peroxide ions, is the rate-determining step [10,11] because it requires dissociation of the O-O bond. If a catalyst that enables easy dissociation of the O-O bond can be developed, the rate of ORR can be improved, even at lower temperatures and the cell performance can be enhanced. To enhance the cathode performance, we studied cathodes coated with catalysts such as lanthanum strontium cobalt ferrite (LSCF) [13], gadolinium strontium cobaltite (GSC) [14], and Ag [15]. LSCF and GSC are known to dissociate oxygen molecules readily because they have many oxygen vacancies. Accordingly, our results indicate that coating the cathode with LSCF or GSC catalysts dramatically reduces cathode polarization [13,14]. Ag is also known as an effective catalyst for O2 adsorption and dissociation; therefore, coating Ag on the cathode also enhances the cell performance [15]. The development of new catalysts that can easily dissociate oxygen dissolved in molten melts to form oxygen ions is one of the most important aspects of enabling long-term operation of MCFCs without degradation of cell performance.
Among the several novel catalyst-coated cathodes that were tested in our laboratory, the Cu-coated cathode demonstrated the highest cell performance; however, the high cell performance did not persist for an extended period. Thus, in the present study, we focus on the Cu-coated cathode and its electrochemical and physical behavior. This study can provide concrete evidence to elucidate the performance-enhancing mechanism of catalyst-coated cathodes and aid in the development of a new cathode for MCFCs.
2. Experimental
A Cu-coated cathode was fabricated by coating Cu powder (Nanotechnology, Inc.) in a 100 nm-thick layer with a spherical shape on a porous Ni plate, which was converted in situ into a lithiated NiO cathode in the cell. The porous Ni plate was coated with the Cu powder suspension via the vacuum suction method as follows: 3 wt% Cu powder was dispersed into ethanol by ultrasonication for 1 h with disperbyk-190 (Daehan, Inc.) added as a dispersant. The prepared Cu suspension was slowly dropped onto the Ni plate, which was placed on a Buchner funnel connected to a vacuum pump. The coated Ni plate was then dried in an oven at 80°C for 1 h. The morphology of the coated cathode was examined via scanning electron microscopy (SEM; NOVA NanoSEM200).
The performances of the Cu-coated cathode and uncoated conventional cathode were evaluated and compared in a single cell with an active electrode area of 100 cm2. Except for the cathodes, the cells were identical in their components and included a Ni-Al anode, Li/K carbonate electrolyte, and LiAlO2 matrix, as described in our previous publications [13-16]. The single cells were operated at 620°C. The gas compositions in the cathode and anode were air:CO2 = 7:3 and H2:H2O:CO2 = 72:18:10, respectively. The single cells were loaded with DC current using an electric loader (ESL300Z, ELTO DC Electronics Co.) and operated at a current density of 150 mAcm−2; the cell performance was measured periodically. To analyze the electrode polarization, electrochemical impedance analysis (EIS) was performed using a Solartron S11287 electrochemical interface and a 1255B frequency response analyzer.
To elucidate the enhanced cell performance of the Cu-coated cathode at the initial stage of operation, the surface of the cathode after 0 h, 24 h, and 150 h of operation was examined by X-ray photoelectron spectroscopy (XPS). A PHI 5000 VersaProbe (ULVAC-PHI, Chigasaki, Japan) system with a microfocused Al X-ray beam (100 μm, 25 W) and a C ls reference at 284.6 eV were used. To understand the change in the state of Cu in the cell during pretreatment, the phase structures of CuO after heat-treatment for 10 h, 20 h, and 30 h at 600°C in a CO2 atmosphere were analyzed by X-ray diffraction (XRD) using a Rigaku D/Max-2400 diffractometer with Cu-Kα radiation.
3. Results and Discussion
In our previous study [13-15], we tested different catalyst-coated cathodes including GSC-, LSCF-, and Ag-coated cathodes. The current density-voltage (J-V) curves of the single cells using these cathodes are presented in Fig. 1 along with the J-V curve of the cell with a Cu-coated cathode. As indicated in this figure, the performance of the Cu-coated cathode, i.e., 0.87 V at a current density of 150 mAcm−2 and 620°C, was significantly better than that of the cathodes coated with LSCF, GSC, or Ag, which are known to be effective catalysts for O2 dissociation. The high voltage of the cell with a Cu-coated cathode at the low operation temperature was unexpected.
Fig. 1.
J-V curves of single cells using uncoated cathode, Cu-coated cathode, LSCF-coated cathode, GSC-coated cathode, and Ag-coated cathode operated at 620°C.

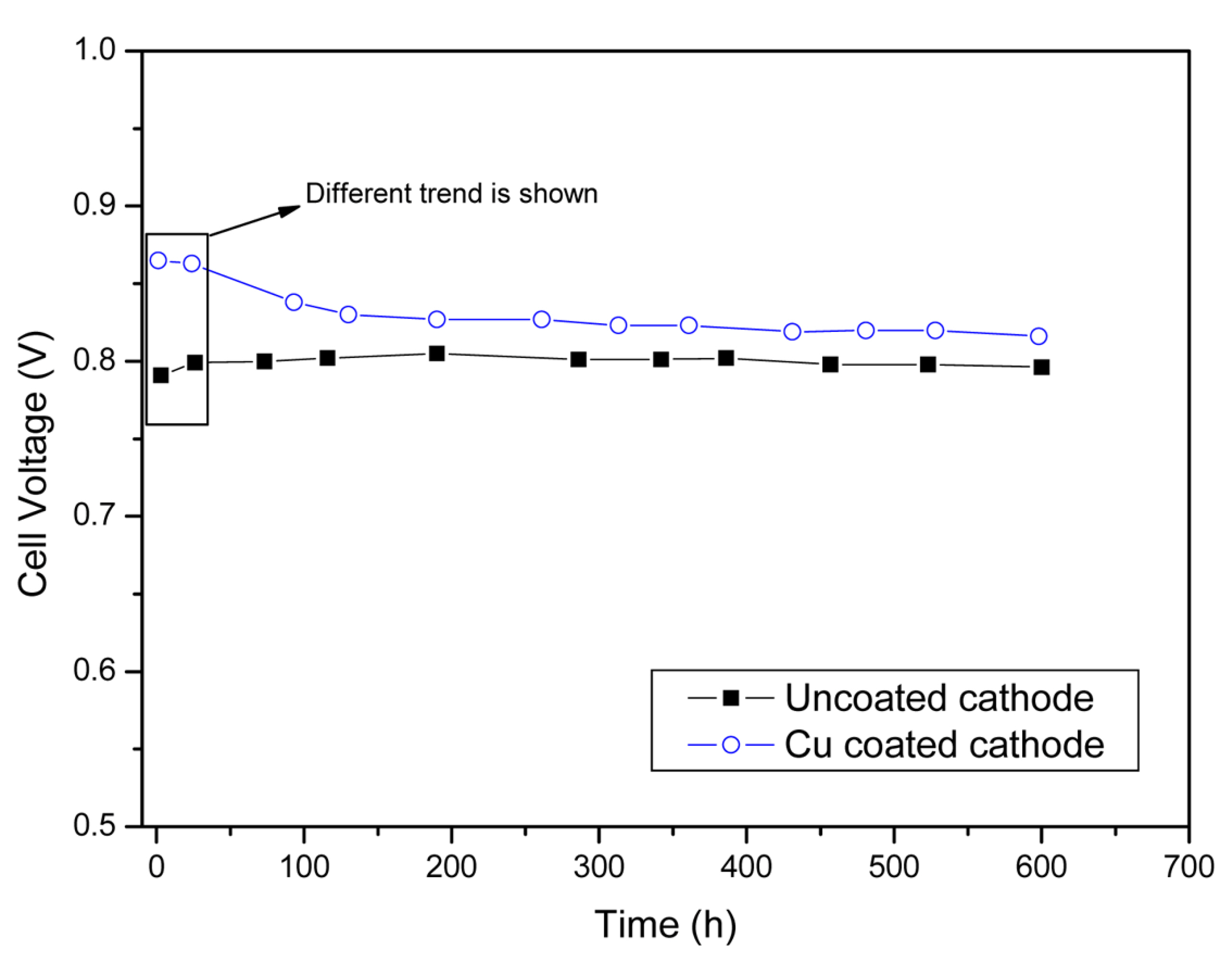
The cell with the Cu-coated cathode was then operated for more than 600 h to examine its long-term stability. Fig. 2 indicates the variation in the voltage of this cell compared with that of a cell with an uncoated cathode. The cell voltages were measured at 620°C at a current density of 150 mAcm−2. As indicated in this figure, the single cell with a Cu-coated cathode had a considerably greater initial voltage (0.87 V) compared to the uncoated cathode. However, the cell voltage rapidly decreased to 0.81 V after 100 h and remained constant at this value for more than 500 h. Conversely, the cell with the conventional uncoated cathode had a stable voltage for more than 600 h of operation. The initial cell voltage was 0.78 V, which increased to 0.8 V after approximately 50-100 h because of electrolyte redistribution in the pores of the components [17]. From this perspective, the behavior of the voltage of the cell with a Cu-coated cathode differed significantly from that of the conventional MCFC single cell. Because the single cells and operation conditions were identical except for the cathodes, the difference in the initial cell performance can be attributed to the different cathodes used.
Fig. 2.
Cell performances of single cells using uncoated cathode and Cu-coated cathode with operating time at current density of 150 mAcm−2 at 620°C.
To understand the effect of the copper coating on changes in cell voltage over time, EIS analysis was performed on single cells with Cu-coated and uncoated cathodes. Fig. 3 displays the Nyquist plot from EIS analysis of single cells using an uncoated cathode and a Cu-coated cathode at 620°C with operation time. In general, the high-frequency semicircle represents the resistance related to the electrochemical reaction and the low-frequency semicircle indicates the resistance related to the mass transfer [18]. Further, we can extract the high-frequency resistance (Rhf) and low-frequency resistance (Rlf) from the EIS data using equivalent circuits [13]. Comparing the Rhf and Rlf values of the cells with Cu-coated and uncoated cathodes, we can determine whether the influence of the Cu coating results from improvements in the electrochemical reaction or mass transfer. As indicated in Table 1, which displays the resistances obtained at different operation times for the two single cells, there is no significant difference in the Rlf values of the two cells with cell operation time. However, the Rhf behaviors of the two cells with time are considerably different. The Rhf values of the cell with a Cu-coated cathode at 2 h, 24 h, 190 h, and 600 h of operating time were 0.29, 0.37, 0.54, and 0.60 Ωcm−2, respectively, whereas those of the cell with an uncoated cathode were 0.67, 0.66, 0.65, and 0.66 Ωcm−2, respectively. The Rhf of the cell with a Cu-coated cathode increased dramatically with operation time, whereas the Rhf of the cell with an uncoated cathode remained essentially constant. Although the Rhf value obtained from the Nyquist plot includes the resistances of the cathode and anode reactions, we can assume that the difference between the Rhf values of the cells with Cu-coated and uncoated cathodes results from the cathode reaction. Because the anodes and operation conditions for each single cell were identical, the anodic Rhf can be assumed to be the same in both single cells. Consequently, it is clear that the Cu coating enhances the initial cell performance by improving the electrochemical reaction on a cathode.
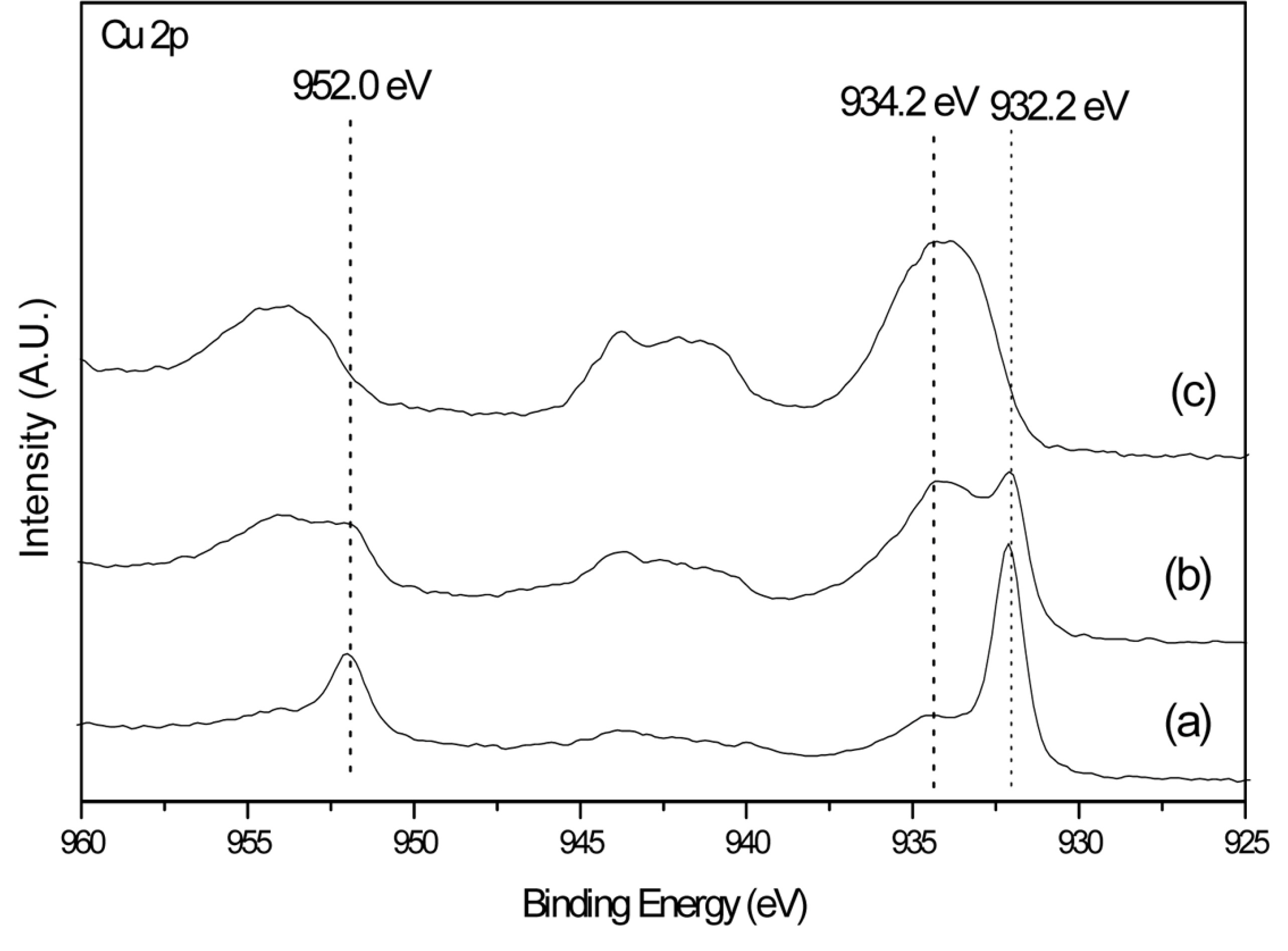
Unfortunately, the Cu coating does not maintain long-term high performance. As illustrated in Fig. 2 and Table 1, the performance of the cell with a Cu-coated cathode decreased and the Rhf was greater during the initial cell operation. It is necessary to elucidate the reason why the cell with a Cu-coated cathode could not maintain a high performance to understand the source of the improved cathode polarization. Because the different cell voltage behavior occurs because of the Cu coating on the cathode and changes with operation time, we focused on investigating the surfaces of the Cu-coated cathodes in single cells operated for different lengths of time via XPS analysis. Interestingly, the XPS data revealed that only the speciation of Cu changed with the operation time and that the Ni and Li states in the lithiated NiO cathode were unaffected by operation time, unlike the case of the uncoated cathode. Fig. 3 displays the XPS data for Cu 2p in the Cu-coated cathodes in a single cell that was operated for 0 h, 24 h, and 150 h. As indicated in this figure, a sharp peak is located at a binding energy of 932.2 eV in the sample with 0 h of operation, whereas two peaks are observed at binding energies of 932.2 and 934.2 eV in the sample with 24 h of operation. After 150 h of operation, a broad peak at 934.2 eV is observed. Therefore, the main peak shifts from 932.2 eV to 934.2 eV with operation time. It is known that the peaks at binding energies of 932.2 eV and 934.2 eV correspond to the 2p/1/2 energy level of Cu+ in Cu2O [19] and 2p/1/2 energy level of Cu2+ in CuO [20], respectively. This indicates that Cu on the cathode is in the form of Cu2O at the initial stage of cell operation and changes to CuO with operating time. As indicated in the cell voltage curve in Fig. 2, the voltage of the single cell with a Cu-coated cathode decreased rapidly after 100 h of operation. We can confidently propose that the change in the cell voltage behavior with the operating time of the cell with a Cu-coated cathode, as displayed in Fig. 2, results from the change in the speciation of Cu on the cathode. Apparently, Cu2O on the cathode significantly enhances the cell performance. However, CuO, which is generated from Cu2O during operation of the cell, does not enhance cell performance.
Fig. 3.
Nyquist plot from EIS analysis of single cells using (a) an uncoated cathode and (b) a Cu-coated cathode at 620°C with operation time.
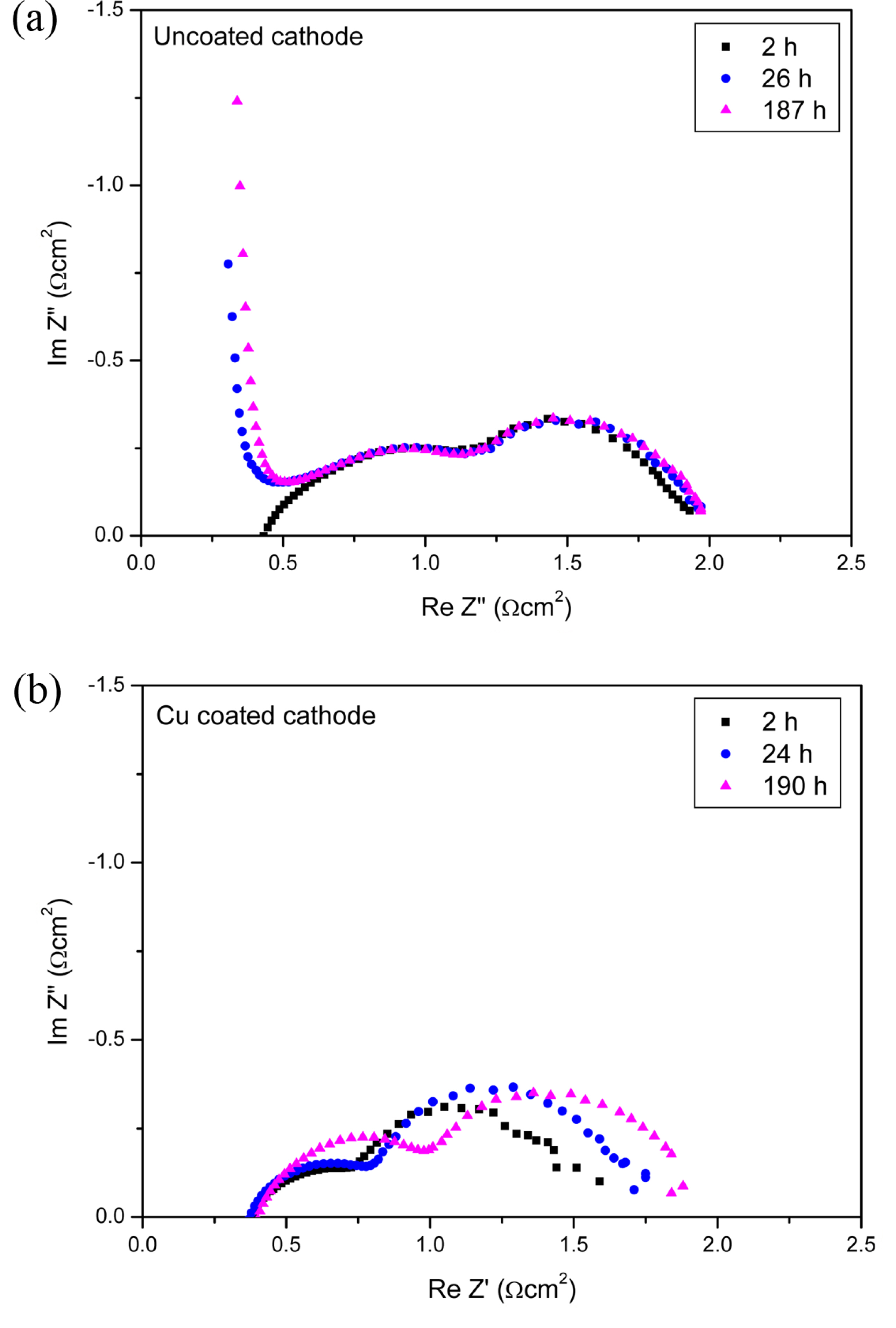
Table 1.
High frequency resistance and low frequency resistance in single cells using uncoated cathode and Cu-coated cathode with operation time.

Cu2O is commonly used as a catalyst for methanol (M) synthesis [21,22], dimethyl carbonate (DMC) synthesis by oxidative carbonylation of methanol [23,24], and CO oxidation [25-28]. Although the details of the catalytic mechanism remain unclear, Cu+ sites are known to be the active sites for M, DMC, and CO oxidation. There are also reports concerning the catalytic activity of Cu2O on O2 dissociation [28,29]. In particular, Zhang et al. reported theoretically calculated results on the adsorption and dissociation of O2 on the surface of Cu2O because this process is important for many types of heterogeneous catalysis [29]. They calculated the adsorption energy and dissociation energy of O2 on perfect and deficient Cu2O (111) surfaces in several modes. According to their results, molecular O2 is adsorbed in the peroxo form (O22−) on both perfect and deficient Cu2O surfaces and easily dissociates with a low activation energy (~20 kJmol−1), especially from deficient surfaces. The reaction energy of O2 dissociation also changes from highly endothermic (over 500 kJmol−1) to exothermic when a Cu2O catalyst is used. Therefore, Cu2O is a powerful catalyst for O2 dissociation. As mentioned in the introduction, peroxide is one of the forms that oxygen assumes during the ORR process and the rate-determining step of the ORR on the MCFC cathode is the dissociation of the O-O bond of the peroxide or superoxide ion. Therefore, during the initial operation of the cell using a Cu-coated cathode, the peroxide ions dissolved in molten carbonate adsorb freely onto the Cu2O surface and then readily dissociate to oxygen ions. This powerful catalytic activity of Cu2O could be the reason for the improved cathode performance during the initial stage of cell operation.
However, Cu2O is converted into CuO during cell operation owing to oxidization by O2, which is one of the reactant gases. According to Uzunova’s report [30], there is minimal difference between the dissociation energy of oxygen on CuO and NiO. Thus, the catalytic activities of CuO and NiO are expected to be similar, which is likely why the voltage of the single cell with a Cu-coated cathode decreases dramatically and then becomes similar to that of a single cell with an uncoated cathode after Cu2O is converted into CuO.
To elucidate how Cu2O forms from Cu before cell operation, we focused on the pretreatment process. Fig. 4 displays our pretreatment schedule and the state of Cu at each step. In the pretreatment process, air is introduced to the cell up to 450°C to remove the organic materials within the matrix. During this period, Cu is readily oxidized to CuO. Upon attaining 450°C, only CO2 flows in the cell to facilitate carbonate melting. Because an oxidizing agent is not introduced between 450 and 620°C, CuO could be reduced. According to the phase diagram [31] of the Cu state and O2 partial pressure, Cu2O exists below an O2 partial pressure of 10−4 atm in the temperature range of 450-650°C. To confirm experimentally if CuO is reduced under CO2, CuO was heat-treated in a CO2 atmosphere at 620°C; the results of the XRD analysis are presented in Fig. 4(b). The intensity of the CuO peak decreases and the intensity of the Cu2O peak increases with increasing heat-treatment time. Therefore, it is confirmed that Cu on the cathode is oxidized to CuO up to 450°C in air, and then CuO reduces to Cu2O between 450-620°C in the CO2 atmosphere during the pretreatment process.
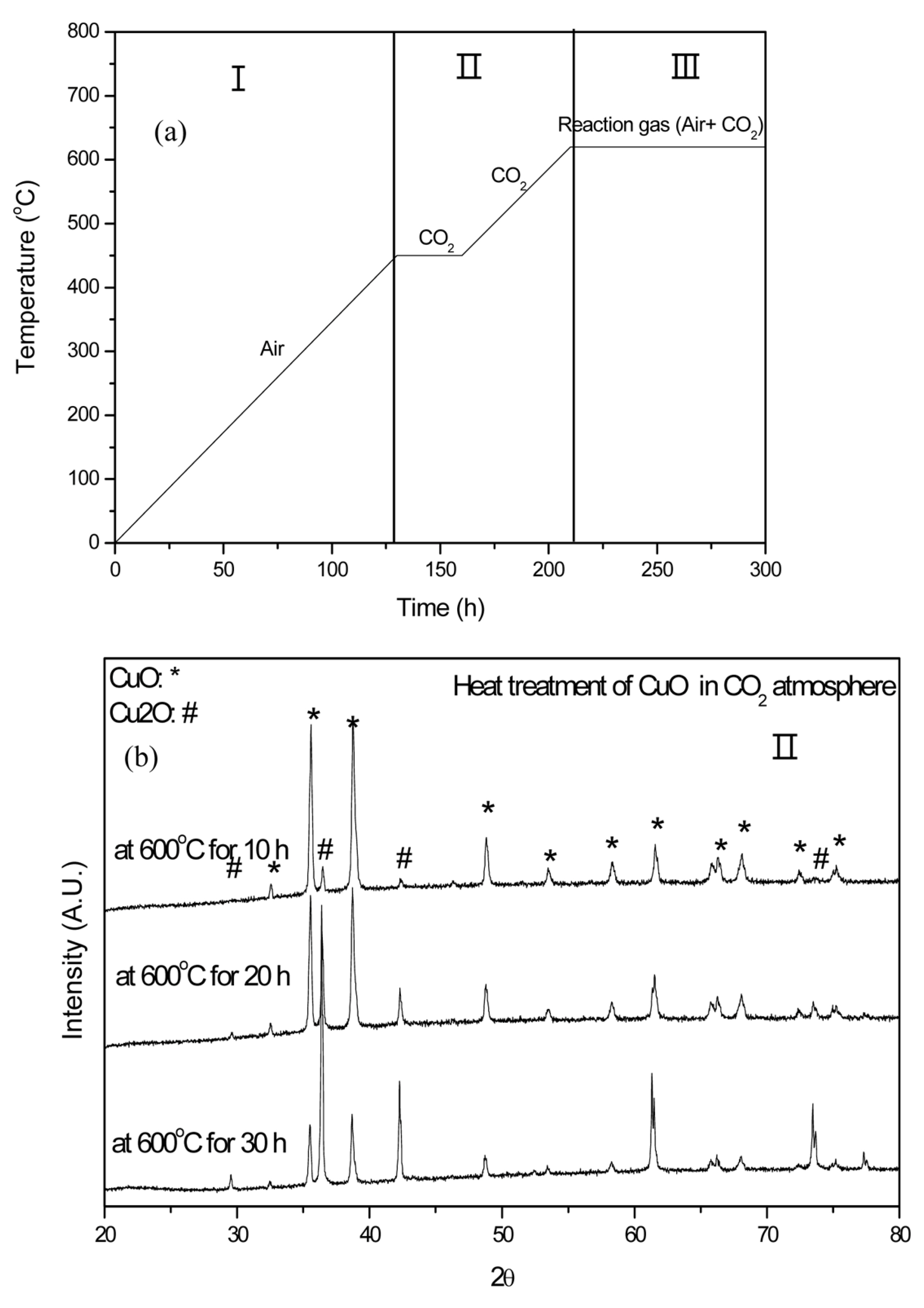
4. Conclusions
In this study, a Cu-coated Ni plate was prepared and evaluated as an MCFC cathode. The cell with a Cu-coated cathode demonstrated a high initial voltage. To verify the mechanism of the enhanced cell performance, several analytical methods were employed; the performance enhancement at the initial stage of cell operation results from the reduction of cathode polarization due to the strong catalytic activity of Cu2O toward O2 dissociation. Unfortunately, the cell with a Cu-coated cathode does not maintain its high voltage for an extended time because Cu2O is oxidized to CuO by O2, which is one of the reactant gases. This study demonstrates that the key to the development of efficient MCFC cathodes is the development of an effective catalyst for dissociation of the O-O bond that is stable in the cathode atmosphere and in carbonate melts.




