


1. Introduction
Lithium-ion batteries are essential power sources for portable electronic devices, electric vehicles, and smart grid storage systems [1-8]. These and other industrial developments have created an increased demand for advanced lithium batteries, which feature a high-performance cathode as the key component [9-15]. Ni-rich compounds, e.g., Li[Ni0.8Co0.15Al0.05]O2, are promising cathode materials with high specific discharge capacity. However, they exhibit several disadvantages such as insufficient cycling performance, rate capability, and thermal stability [16-20]. To address these issues, modification of cathode material surfaces with carbon has been studied [21-25]. Carbon layers exhibit high electronic conductivities. Therefore, the layers compensate for the low conductivities of cathode materials, enhancing their rate capabilities. Moreover, they can also suppress side reactions between cathodes and electrolytes, leading to improved stability [26-30].
In this study, the surface of Li[Ni0.8Co0.15Al0.05]O2 was carbon coated, using polyvinylidene fluoride (PVDF) as a novel organic source. PVDF is generally used as a binder for making electrodes. However, it can be homogeneously coated on the cathode surface using a solvent such as N-methyl-2-pyrrolidone (NMP), acting as a good carbon source with high electric conductivity during carbonization. The carbon-coated Li[Ni0.8Co0.15Al0.05]O2 cathode was expected to show an improved rate capability owing to the good conductivity of the carbon layer.
2. Experimental Section
Pristine Li[Ni0.8Co0.15Al0.05]O2 powder was sourced from ECOPRO, and its surface was modified using PVDF (Sigma-Aldrich) as a carbon source (PVDF contents of 0.5, 1.0, and 3 wt.%). To produce the coating solution, PVDF was dissolved in NMP (Sigma-Aldrich) at 70°C for over 24 h. This solution was treated with pristine Li[Ni0.8Co0.15Al0.05]O2 and stirred at 70°C to achieve complete solvent evaporation. The obtained sample was dried in a vacuum oven at 90°C for 24 h and carbonized at 400°C for 2 h in N2 atmosphere. X-ray diffraction (XRD) patterns of the prepared powders were recorded in a 2θ range of 10-90° using monochromatized Cu Ka radiation (λ = 1.5406 Å) (Rigaku MiniFlex II, Rigaku). The sample surface morphology was analyzed by transmission electron microscopy (TEM, JEOL-4010), while the carbon content of carbon-coated Li[Ni0.8Co0.15Al0.05]O2 samples was determined using thermogravimetric analysis (TGA, Q500 V20.13 Build 39) involving heating to 550°C under atmospheric conditions. For electrochemical testing, a cathode slurry was prepared by mixing Li[Ni0.8Co0.15Al0.05]O2 (pristine and carbon-coated), carbon black (Super P), and PVDF in a weight ratio of 80:10:10, respectively. The components were ball-milled for homogeneous mixing, coated on aluminum foil, and dried at 90°C for 2 h. The fabricated 2032 coin-type cells contained the cathode, separator, Li-metal anode, and electrolyte (1 M LiPF6 in ethylene carbonate/dimethyl carbonate (1:1 v/v)) components. The cathode loading weight and thickness were 5-6 mg·cm−2 and 40-50 μm, respectively. The fabricated cells were galvanostatically cycled (WonATech voltammetry system) in a potential range of 4.5-3.0 V at various charge-discharge rates. Impedance measurements were performed by applying AC voltage (5 mV amplitude, 0.1 Hz-100 kHz frequency) using an electrochemical workstation (AMETEK, VersaSTAT 3). The thermal stability of the fully charged electrode (4.5 V) was analyzed by differential scanning calorimetry (DSC, Mettler Toledo). The cathode material (7 mg) was mixed with the electrolyte and sealed in a high-pressure DSC pan. A heating rate of 10°C/min and a temperature range of 25-350°C were used for the DSC tests.
3. Results and Discussion
Fig. 1 shows TEM images of pristine and carboncoated Li[Ni0.8Co0.15Al0.05]O2 powders. Hereafter, we refer to the carbon-coated Li[Ni0.8Co0.15Al0.05]O2 samples prepared using varying amount of PVDF as 0.5, 1.0, and 3.0 wt.% as the carbon source as 0.5, 1.0, and 3.0 wt.% PVDF samples, respectively. Li[Ni0.8Co0.15Al0.05]O2 powders consisted of spherical particles with diameters of 5-6 μm. As shown in Fig. 1a, these particles displayed a smooth surface without other heterogeneous surface layers. In contrast, the surface of the carbon-coated Li[Ni0.8Co0.15Al0.05]O2 cathodes was covered with a layer of film, corresponding to the carbon layer prepared by PVDF carbonization. The thickness of the above layer increased with increasing PVDF content.
Fig. 1.
TEM images of Li[Ni0.8Co0.15Al0.05]O2 samples. (a) Pristine sample, (b) 0.5 wt.% PVDF sample, (c) 1.0 wt.% PVDF sample, and (d) 3.0 wt.% PVDF sample.

The amount of PVDF was adjusted to control the carbon content and/or the carbon layer thickness on the cathode surface. However, the actual carbon content of the samples after carbonization had to be measured by other methods. Therefore, the pristine and carbon-coated Li[Ni0.8Co0.15Al0.05]O2 powders (after heating at 400°C) were subjected to TGA. As shown in Fig. 2, the 0.5, 1.0, and 3.0 wt.% PVDF samples exhibited weight losses of ~0.16, 0.3, and 1.4 wt.% at 550°C, respectively. These weight losses are attributed to the evaporation of carbon during thermal treatment, corresponding to the sample carbon content. The carbon coating formed from the organic precursor can affect the crystal structure of the cathode, since carbonization is accompanied by reactions with oxygen. The XRD patterns of pristine and coated samples were compared to determine the phase integrity after carbon coating (Fig. 3a). No significant differences were observed between pristine and carbon-coated samples, with their patterns corresponding to the general α-NaFeO2 structure (R-3m space group). Figs. 3b-d show the enlarged narrow-angle XRD peaks of Fig. 3a for further detailed examination. The peaks of pristine and carbon-coated samples were almost identical, indicating that the sample phase did not change significantly during the carbon coating process.
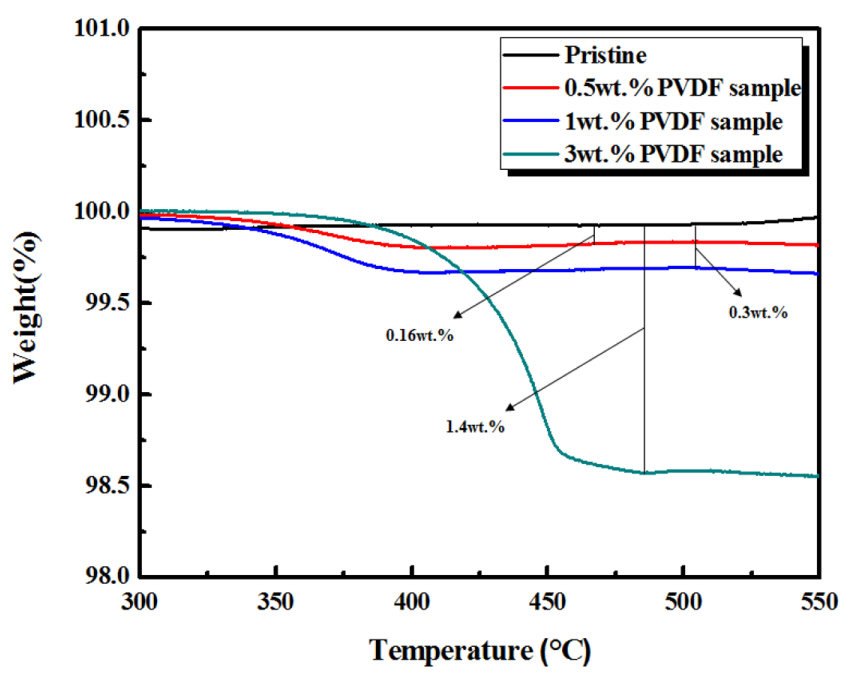
Fig. 3.
XRD patterns of pristine and carbon-coated samples. (a) Full range (10-90), (b) 18-20°, (c) 35-50°, and (d) 58-70°.

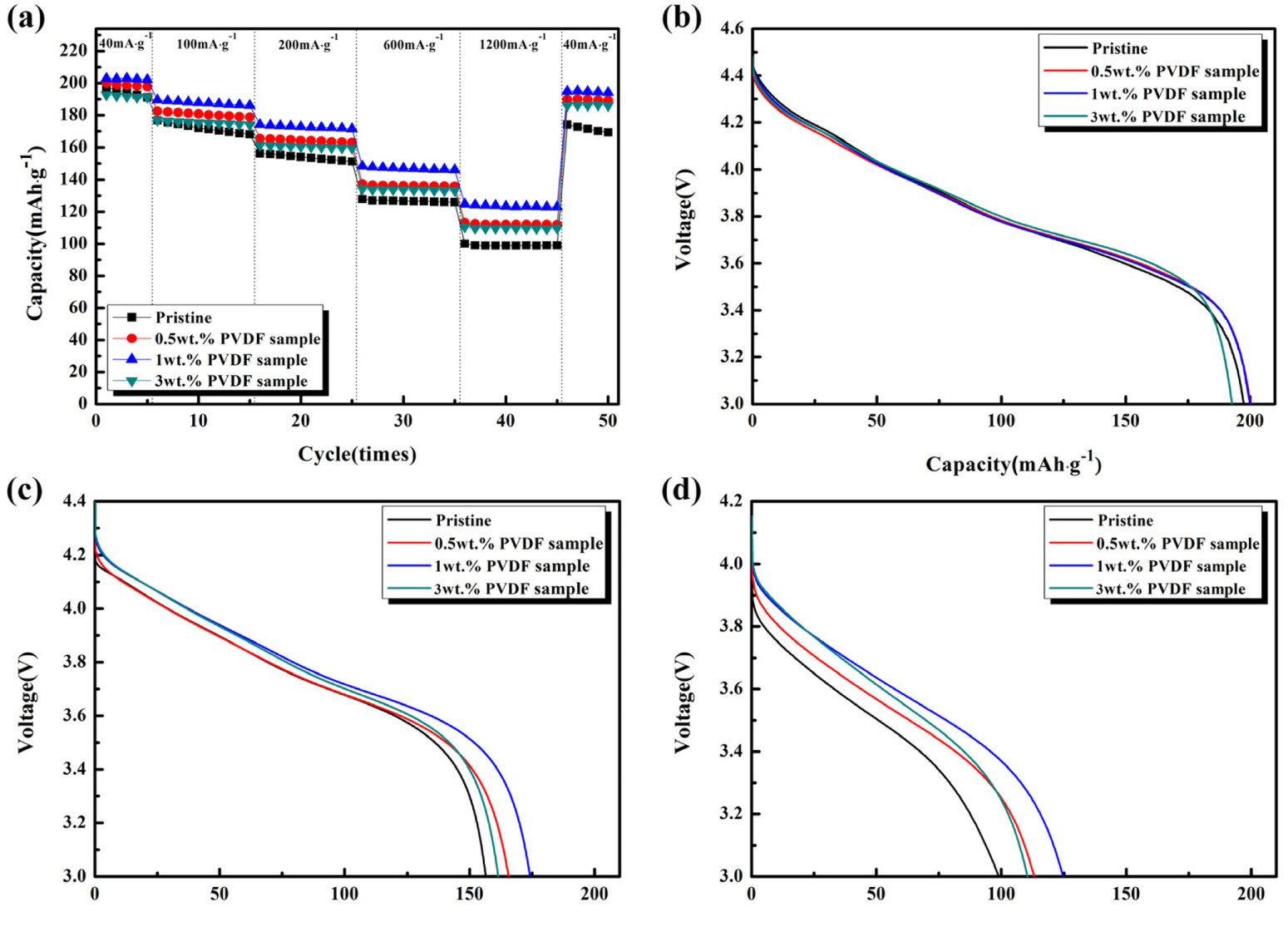
The electrochemical properties of all samples were characterized to determine the effect of the PVDF-derived carbon layer. Fig. 4a shows the discharge capacities of pristine and carbon-coated Li[Ni0.8Co0.15Al0.05]O2 cathodes at various current densities (40, 100, 200, 600, and 1200 mA·g−1) in a voltage range of 4.5-3.0 V. At a current density of 40 mA·g−1, the pristine and carbon-coated samples exhibited similar discharge capacities. However, as the current density increased to 1200 mA·g−1, the carbon-coated samples showed higher discharge capacities than the pristine sample, suggesting that their rate capability was enhanced by introducing the carbon coating. Among the samples tested, the best rate capability was shown by the 1.0 wt.% PVDF sample. Figs. 4b-d show the voltage profiles of pristine and carbon-coated samples measured at current densities of 40, 200, and 1200 mA·g−1, respectively. As shown in Fig. 4b, the sample discharge capacities were around 190-200 mAh·g−1 at 40 mA·g−1. A slight difference of up to 10 mAh·g−1 was observed between the capacities of pristine and carbon-coated samples. However, this difference increased as the current density was raised to 1200 mA·g−1. Under these conditions, the capacity of the pristine sample was only ~100 mAh·g−1, while that of the 1.0 wt.% PVDF sample was ~125 mAh·g−1. The enhanced rate capability of the carbon-coated sample is attributed to the surface carbon layer with high electronic conductivity. However, despite its higher carbon content, the 3.0 wt.% PVDF sample showed a rate capability inferior to that of the 1.0 wt.% PVDF sample, suggesting that the carbon layer of the former was too thick, resulting in adverse effects such as deterioration of surface layer phase integrity and/or interruption of the Li-ion movement.
Fig. 4.
(a) Discharge capacities of pristine, 0.5 wt.%, 1 wt.%, and 3 wt.% PVDF samples at current densities of 40, 100, 200, 600, and 1200 mA·g−1 in a voltage range of 4.5-3.0 V. Sample discharge profiles at (b) 40 (c) 200, and (d) 1200 mA·g−1.
The sample impedance spectra confirm the effect of the carbon coating layer. The size of the semicircle in Nyquist plots represents the impedance related to the charge transfer and the solid-electrolyte interface. As shown in Fig. 5, the semicircle obtained for the carbon-coated sample was smaller than that obtained for the pristine sample, indicating that the impedance decreased because of the carbon coating layer. The semicircle obtained for the 1.0 wt.% PVDF sample was smaller than that obtained for other samples, explaining the better rate capability of the former (Fig. 4).
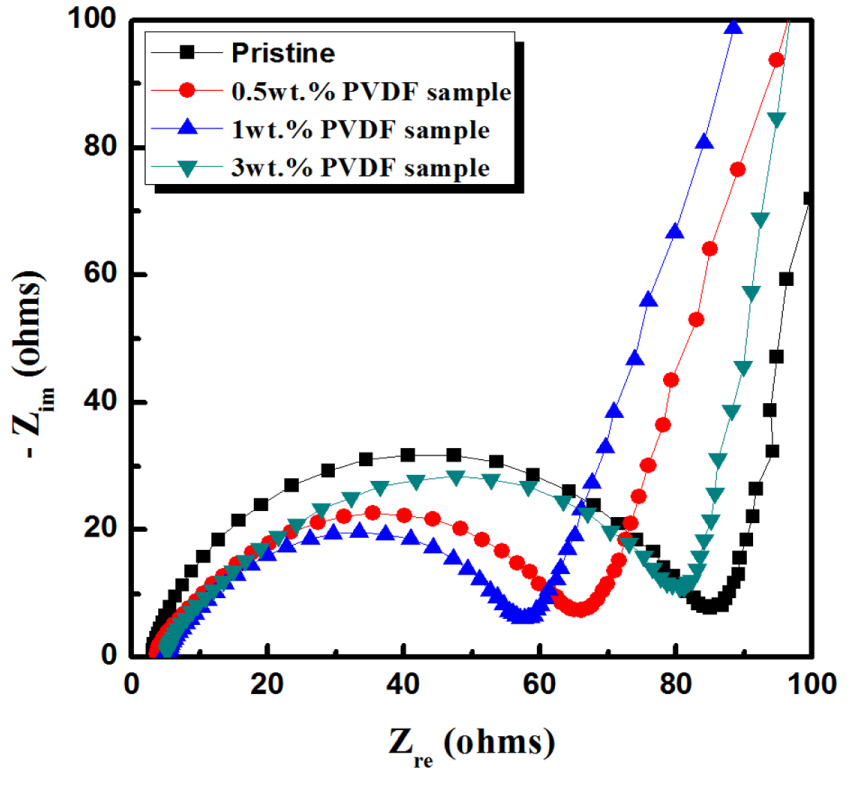
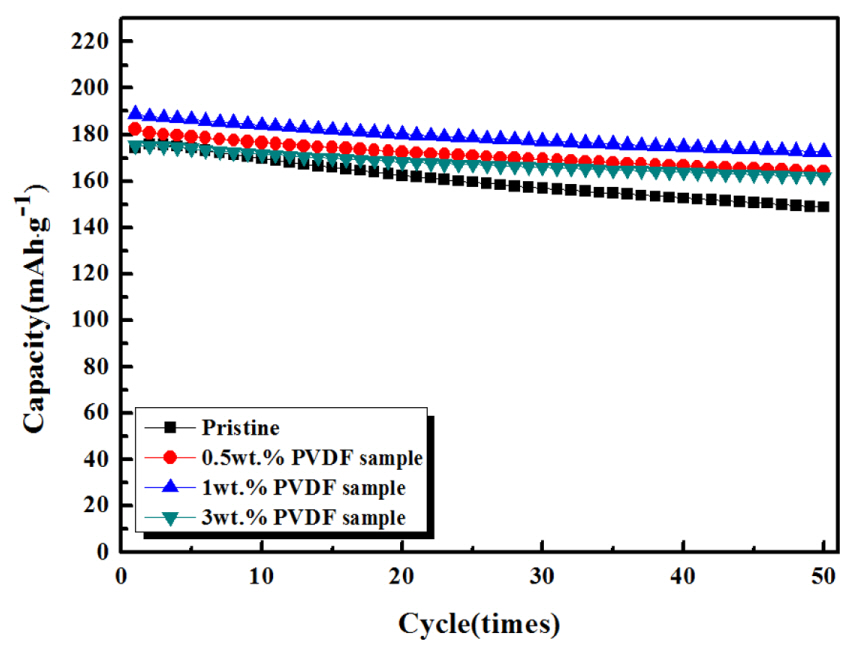
Fig. 6 shows the sample cycling lifetime at a current density of 100 mA·g−1. The carbon-coated samples showed somewhat higher capacities than the pristine one, attributed to the superior rate capability of the former. The capacity of all samples gradually decreased during cycling because of side reactions such as the dissolution of transition metals. However, the carbon-coated samples exhibited a smaller capacity loss than the pristine sample, indicating the superior cycling performance of the former. The pristine sample showed a capacity retention of ~85.4% over 50 cycles, while 0.5, 1.0, and 3.0 wt.% PVDF samples showed capacity retention values of ~90.1, 91.4, and 92.3%, respectively. This result implies that the carbon layer prepared from the PVDF source can suppress side reactions between the cathode and the electrolyte and increase cathode stability during cycling.
Fig. 6.
Cycling performances of pristine, 0.5 wt.%, 1 wt.%, and 3 wt.% PVDF samples at a current density of 100 mA·g−1.
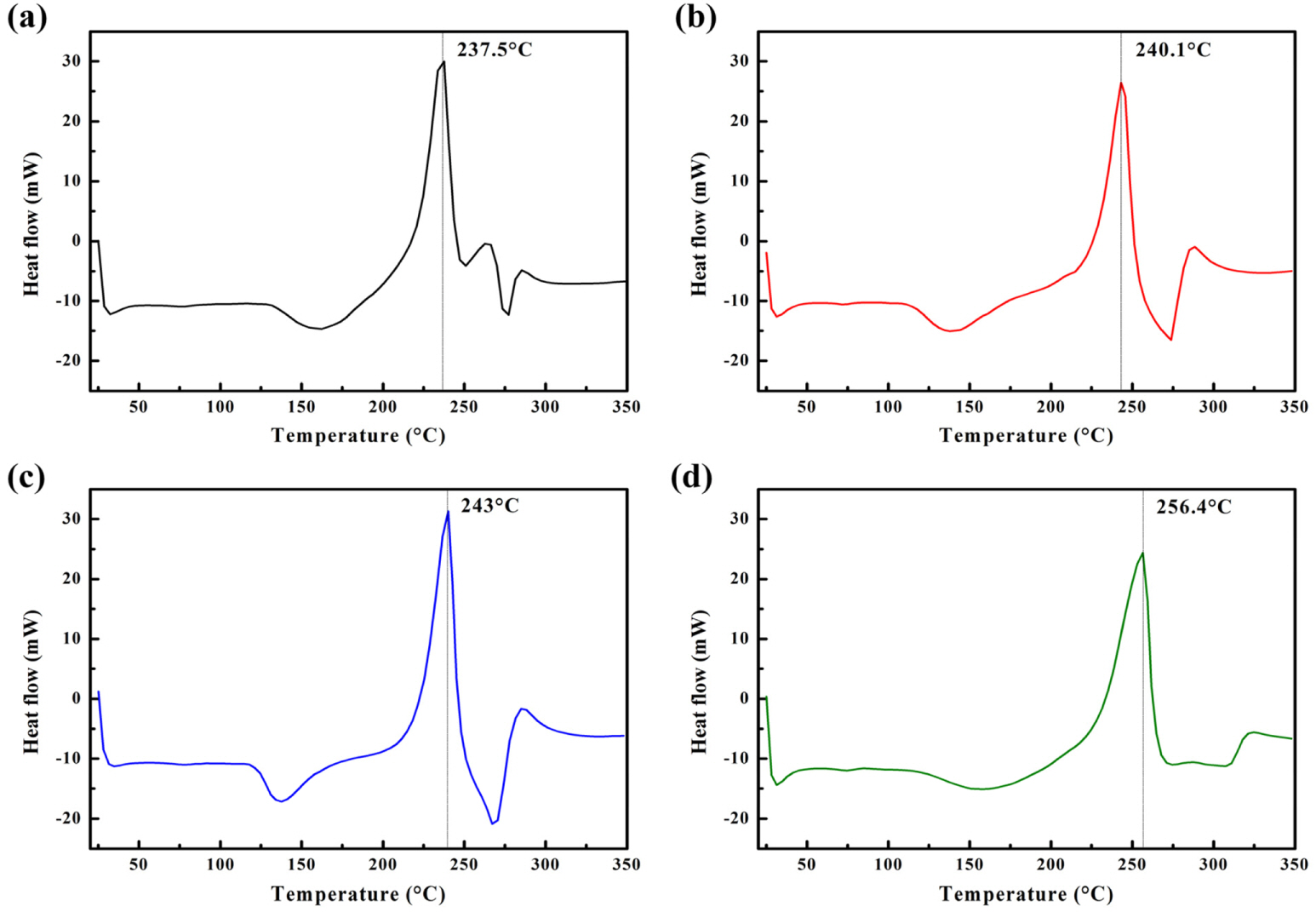
The thermal stability of pristine and carbon-coated samples was also examined to investigate the effect of the carbon coating. As shown in Fig. 7a, an intense exothermic peak was observed at ~273°C for the pristine sample, progressively shifting to higher temperatures with increasing carbon content of the coated samples. The peak intensity for the carbon coated samples also decreased compared to that of the pristine sample. This result confirms that the thermal stability of the Li[Ni0.8Co0.15Al0.05]O2 cathode could be improved by the carbon coating layer, which suppresses the reaction between the cathode and the electrolyte.
4. Conclusions
The Li[Ni0.8Co0.15Al0.05]O2 cathode was surface-modified by a carbon layer prepared using PVDF as an organic precursor, as confirmed by TEM imaging. The carbon content of 0.5, 1.0, and 3.0 wt.% PVDF samples was 0.16, 0.3, and 1.4 wt.% of the pristine sample, respectively. The XRD patterns of the samples were unchanged after carbon coating, indicating that carbonization did not affect phase integrity. The rate capability of the Li[Ni0.8Co0.15Al0.05]O2 cathode was improved by carbon coating owing to its high electronic conductivity, with the low impedance of the carbon-coated samples (compared to that of the pristine sample) supporting this result. Moreover, the cycling performance and thermal stability of carbon-coated samples were superior to those of the pristine sample, implying that the carbon coating layer also suppressed side reactions between the cathode and the reactive electrolyte, thus enhancing cathode stability.




