


1. Introduction
In recent years, lithium ion batteries (LIBs) have drawn much attention due to their advantages over traditional secondary batteries. The anode material is a very important component for LIBs and can be usually divided into three kinds depending on its type of reactivity upon lithiation process, i.e. intercalation-type (e.g. graphite [1]), alloying-type (e.g. Sn [2], Si [3] and Ge [4]), and conversion-type (e.g. Co3O4 [5], V2O5 [6], Fe2O3 [7] and MoO3 [8]). Although graphite has been extensively employed as anode material in commercial LIBs for many years, its relatively low Li ion storage capacity (theoretical capacity of 372 mAh/g) restricts the further improvement of LIBs. Fortunately, transition metal oxides (TMOs) demonstrate a several times higher capacity as compared to that of graphite because of the multiple electron reaction in conversion process [9]. The high theoretical capacity, high safety and huge abundance make the TMOs to be very promising anode materials [10].
MoO3 is an environmentally friendly TMO that has been paid much attention over the past decades as an anode material owning to its low cost, low toxicity and high theoretical capacity (as high as 1,117 mAh/g) which is about three times higher than that of graphite. However, its huge unit cell volume expansion during the Li ion insertion [11] and poor electronic/ionic conductivity [12] usually reduce its cyclic and rate performance. To overcome these drawbacks, different MoO3 nanostructures have been prepared for enhancing their electrochemical performance. The preparation of MoO3 as nanobelts [12], nanorods [13], nanowires [14] and nanosheets [15,16] has been widely investigated. In addition, it has been confirmed that the electrochemical performance is strongly related to the electrical conductivity of the electrodes. In this regard, different conductive materials, such as conductive polymer [17], graphene [18- 20], amorphous carbon [21-23], carbon nanotubes [24] and In2O3 [8] have been used for incorporating MoO3 within the electrode base material.
In this work, a simple method to fabricate MoO3 and C nanocomposites was proposed. Firstly, polyaniline (PANI) doped with ammonium heptamolybdate was synthesized. Then MoO3 and carbon composite was obtained by post heat treatment of doped PANI. In this synthesis route, the molybdenum source is well dispersed in PANI chains so that the residual carbon can be homogeneously distributed in MoO3/C composite. The electrical conductive property and electrochemical performance of as prepared MoO3/C were significantly enhanced as compared with pristine MoO3. Furthermore, the temperature dependence of Li ion diffusion coefficient (DLi) determined by EIS obeys Arrhenius law. To the best of our knowledge, the relationship between capacity and temperature for MoO3/C has not been reported previously.
2. Experimental Section
Ammonium heptamolybdate tetrahydrate (AMo, (NH4)6Mo7O24·4H2O) and ammonium persulfate (APS, (NH4)2S2O8) are of analytical grade and used as received. Aniline (AN) was distilled under reduced pressure and stored in the refrigerator before being used. AMo-doped polyaniline was synthesized by a method described in the literature [25]. Firstly, 4.0 g of AMo and 2 mL of AN were dissolved in a HCl solution (1 M, 100 mL) under ultrasonication to form solution A. Then 6.2556 g of APS (APS-to-AN molar is 1.25:1) was dissolved in another 100 mL of HCl solution (1 M) to form solution B. Then, solution B was added dropwise into solution A and reacted for 6 h in an ice bath. The precipitate was filtered and washed thoroughly with deionized water and ethanol until the filtrate was colorless. Finally, the product was dried in vacuum at room temperature for 48 h and named as AMo-PANI. For comparison purposes, emeraldine salt (ES) was prepared by the same method without adding AMo.
The AMo-PANI was calcined in a nitrogen atmosphere at 800 ℃ for 3 h, obtaining a black product. Such product was confirmed to be a Mo2C and carbon composite (Mo2C/C). Afterwards, MoO3/C powder was obtained by calcining Mo2C/C in air at 310 ℃ for 1 h. Thermal gravimetric analysis was conducted on a simultaneous thermal analyzer (STA 449F3, Netzsch, Germany) in the temperature range of 40 - 700 ℃ with a heating rate of 10 ℃/min in air. The carbon and absorbed water contents of MoO3/C are 27.1 % and 1.3 % obtained from TGA curve, respectively. In contrast, pristine MoO3 without carbon was obtained by directly calcining AMo-PANI at 500 ℃ in air for 1 h.
The samples were characterized by powder XRD (DX-2700B X-ray diffractometer, Cu Kα, λ = 1.54184 Å, China), FT-IR (Bruker Vertex 70 IR spectrometer, 4000-400 cm−1, Germany) and FE-SEM (SU8010, 3.0 kV, Japan). The conductivities of different samples were measured by four-terminal method described previously [25]. In this measuring system, the 2400-C source measure unit (Tektronix, America) served as current source and the 34401A digital multimeter (Agilent, America) with 6½ digital resolution served as voltage meter. The slope of the voltage vs. current plot is used for determining the resistance (R) of the sample. The conductivity (σ) of the sample can be obtained by σ = l/(d × b × R), where l is the distance between the two middle electrodes, d and b are the width and height of the cuboid sample, respectively. CR2025 coin cells with different anode materials were assembled in an argon-filled glovebox with the moisture and oxygen concentrations kept below 1 ppm. MoO3/C or pristine MoO3 were used as active material and mixed with acetylene black, polyvinylidene difluofide in a 8:1:1 weight ratio. Then the mixture was dissolved in 1-menthyl-2-pyrrolidi-none to form a slurry. The working electrode was made by coating the slurry on a Cu foil and dried in vacuum. 1M LiPF6 in a mixture of ethyl carbonate, dimethyl carbonate and diethyl carbonate (1:1:1 in volume) was utilized as the electrolyte and a porous polypropylene film (Celgard 2400, Celgard, Inc., USA) served as separator. Li foil was used as the counter electrode in the cell. Galvanostatic charge/discharge measurements were carried out on a cell tester (CT2001A, LAND electronics, China) in the voltage range of 0.01 - 3.0 V vs. Li/Li+. The current density and specific capacities of MoO3/C are calculated based on the mass of molybdenum trioxide. Cyclic voltammetry (CV) and electrochemical impedance spectrum (EIS) were performed on an electrochemical workstation (CHI 660E, CH Instruments, China). The EIS test was measured by applying a DC bias with its value equal to open circuit voltage of the cells and an AC oscillation of 5 mV over the frequencies from 100 kHz to 1 mHz. All the electrochemical tests were carried out at constant and fixed temperature. The working temperature from 20 ℃ to 60 ℃ was controlled by the PID algorithmic program, while the electrochemical tests at 0 ℃ were conducted in an ice bath.
3. Results and Discussion
3.1 Materials Characterization
The XRD patterns of the AMo-PANI, ES and AMo are shown in Fig. 1a. The XRD pattern of ES exhibits a typical partially crystalline polymeric diffraction feature between 20 ° and 30 °, whereas AMo-PANI shows an additional diffraction peak around 7.5 ° The peaks appearing within the small diffraction angle for PANI doped with different acids have been previously observed by many researchers: for example, the diffraction peaks at 6.4 ° and 6.7 ° correspond to the doping of cardanol [26] and 12-tungstophosphoric acid [27], respectively. For our samples, it is believed that the peak near 7.5 ° is related to the existence of molybdate counter-ions in PANI, since the XRD pattern of ES does not show any peak around this position. It should also be noted that no diffraction peak corresponding to AMo crystal can be found in the XRD pattern of AMo-PANI, suggesting that AMo anions are homogeneously dispersed in PANI due to the electrostatic driving force towards neutrality between PANI chains and AMo anions (see Scheme 1).
Fig. 1.
(a) XRD patterns of AMo-PANI (red), ES (blue) and AMo (black); (b) XRD patterns of Mo2C/C, MoO3/C and pristine MoO3.
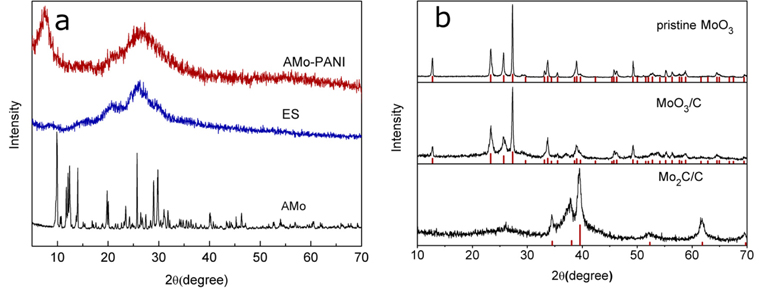
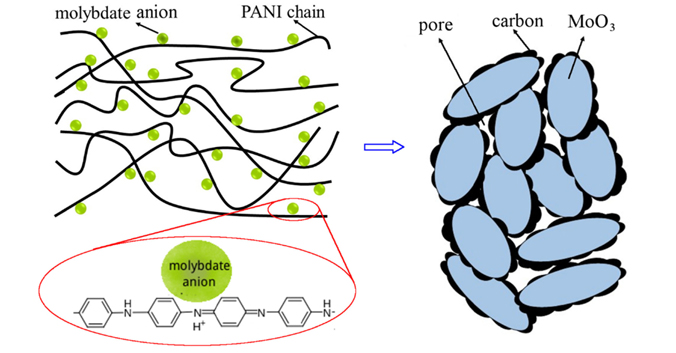
Fig. 1b shows the XRD patterns of Mo2C/C, MoO3/C and pristine MoO3, respectively. The XRD pattern of Mo2C/C is indexed to Hexagonal Mo2C (JCPDS: 65-8766), and those of MoO3/C and MoO3 are indexed to orthorhombic MoO3 (JCPDS: 05-0508). A broad weak peak between 20° and 30° only emerges in the XRD patterns of Mo2C/C and MoO3/C which cannot be found in the pattern of pristine MoO3. It is believed that this peak derives from the amorphous carbon pyrolyzed from the PANI chains. Moreover, it is known that AMo can be decomposed to MoO3 around 400 ℃ at ambient pressure [20,23]; concomitantly, we have found that the MoO3 and C composite can be obtained by calcining AMo-PANI at 400 ℃ under N2 atmosphere but it portrays a poor conductive behavior (ca. 10−5 S/cm). For enhancing the electrical conductivity of the composite, AMo-PANI was firstly calcined at a higher temperature of 800 ℃ under N2 atmosphere to obtain Mo2C/C (Fig. 1b); upon oxidation of Mo2C/C at 310 ℃ in air, a MoO3 and C composite with excellent electrical conductivity is obtained (1.5 S/cm). Thus, it is expected that the latter composite with higher electronic conductivity is a good candidate as electrode material.
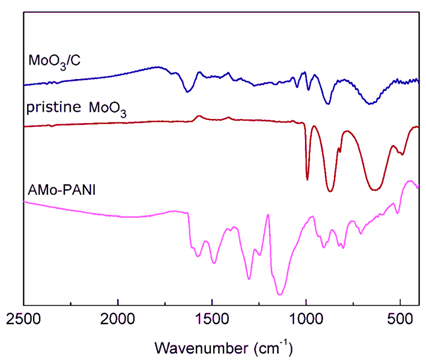
The FT-IR spectra of AMo-PANI, MoO3/C and pristine MoO3 are shown in Fig. 2. typical absorption bands of PANI can be observed from the FT-IR spectrum of AMo-PANI. For example, the band located at 1143 cm−1 is due to the quinonoid unit of doped PANI. The bands located at 1248 cm−1 and 1302 cm−1 are assigned to C-N stretching of the benzenoid unit, while the bands at 1494 cm−1 and 1574 cm−1 are assigned to C=C stretching of quinonoid and C=N stretchings of benzenoid units, respectively [28]. The pristine MoO3 shows three main fingerprint vibrational modes of crystalline MoO3 between 500 - 1000 cm−1. The bands located at 648 cm−1, 881 cm−1 and 987 cm−1 are attributed to the bending of Mo-O-Mo units and the vibrations of the Mo-O and terminal Mo=O moieties, respectively [29]. In the FT-IR spectrum of MoO3/C, the characteristic absorption bands for MoO3 can be clearly observed. The absorption bands located at 1047 cm−1 and 1639 cm-−1 are attributed to C=C and C-O moieties [30], which only appeared in the spectrum of MoO3/C. This evidence proves that carbon is successfully incorporated within MoO3.
The FE-SEM images of MoO3/C and pristine MoO3 are shown in Fig. 3. It can be observed that pristine MoO3 presents a granular morphology (Fig. 3a) that clearly exhibits the layered nature of MoO3. The size of the granules lies in the sub-micrometer scale (500-1000 nm), although a few of them have a size of 20-50 nm. The SEM image shown in Fig. 3b indicates that the MoO3/C sample also has a similar granule morphology, their size falling in the 100-250 nm range, which is smaller than that of pristine MoO3, evincing that the growth of granules is restricted by the amorphous carbon. The porous granules in MoO3/C can be seen from the higher magnification FESEM image shown in the inset in Fig. 3b, which may be related to the decomposition of PANI during the calcination process and possibly benefit their electrochemical performances. Similar micrographs of the porous structure for other TMOs and carbon nanocomposites were also previously reported [23,31].

3.2 Eletrochemical Characterization
The electrochemical Li-storage properties of MoO3/C and pristine MoO3 were studied by CV and galvanostatic charge/discharge measurements. Two pairs of redox peaks around 1.3 V and 0.3 V for MoO3/C and pristine MoO3 can be observed from CV curves (Fig. 4a). These redox peaks reflect the two stages of the lithiation/delithiation process in MoO3 [32,33] and the corresponding redox reactions can be described with the following reactions:
Fig. 4.
(a) CV curves, (b) cyclic performance at 0.5 A/g (initial cycle is measured at 0.1 A/g), (c) EIS plots (inset show the EIS plots of EIS with larger magnification) and (d) rate performance of MoO3/C and pristine MoO3.

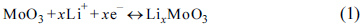
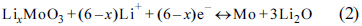
The first stage (Eqn. (1)) is assigned to the insertion of Li ions into the structure of MoO3 to form LixMoO3. The second stage (Eqn. (2)) revealing a relatively higher specific capacity than the previous stage is ascribed to the Li ions inserting into LixMoO3 to form the metal of Mo and the lithium oxide. A small reduction peak near 0 V can be observed too. It seems that this peak is ascribed to the intrinsic character of MoO3 since similar peak was observed in other literatures [20,29]. The MoO3/C redox peaks are more obvious than that of pristine MoO3. In addition, the integrated area surrounded by CV curve (i.e. charge/charge capacity) of MoO3/C is much larger than that of pristine MoO3, which indicates that the porous structure in MoO3/C enhances the specific surface area and effectively shortens the diffusion lengths of charge carriers, thus facilitating the insert/extraction process of Li ions.
The cycle performance of both MoO3/C and pristine MoO3 at a current density of 0.5 A/g (the initial cycle current density is 0.1 A/g) in the voltage range of 0.01 - 3 V is plotted in Fig. 4b. The initial discharge capacities of MoO3/C and pristine MoO3 are found to be 1922.5 mAh/g and 1422.0 mAh/g and then decreased to 991.5 mAh/g and 406.6 mAh/g , respectively. Likewise other TMO anodes [12,31], the capacity loss in the second cycle is mainly due to the irreversible decomposition of electrolyte as well as the formation of solid electrolyte interface (SEI) layer. The discharge capacity for MoO3/C is retained at 608.0 mAh/g after 100 cycles, corresponding to a 61.3% retention with respect to the second cycle. The capacity of pristine MoO3, however, decreases rapidly from 406.6 mAh/g (2nd cycle) to 115.0 mAh/g (100th cycle), corresponding only to a 28.3 % retention, apparently lower than that of MoO3/C. Obviously, the capacity of MoO3/C has been significantly improved as compared to that of pristine MoO3. In this regard, it has been confirmed that the deterioration of MoO3 cycle performance is mainly due to the volume expansion during the lithiation/delithiation process [18]. For the MoO3/C sample, the amorphous carbon around MoO3 “buffers” the volume expansion/contraction caused by the intercalation/deintercalation reaction, yielding a cycle performance that is much better than that of pristine MoO3.
EIS measurements for both MoO3/C and pristine MoO3 were carried out (Fig. 4d). Two compressed semicircles in high/middle frequencies and an oblique line in low frequency can be clearly observed. As previously reported [34-36], the Ohmic resistance (Rs) of the sample is determined by the intercept in the real axis; the semicircle in the high frequency range reflects the resistance related to the Li ion diffusion across the electrode-electrolyte interface (Rf); and the semicircle in the middle frequency range represents the charge transfer resistance (Rct). The oblique line at low frequencies is attributed to the Warburg impedance, related with the ionic diffusion into the bulk electrodes. As it can be seen, the intercept and diameter of both semicircles for MoO3/C are much less than those for pristine MoO3, indicating that the amorphous carbon enhances the electrical conductivity of MoO3/C as well. In Fig. 4d, the rate performance of MoO3/C measured at current densities from 0.2 A g−1 to 3 A/g−1 is plotted. The mean discharge capacity of MoO3 /C is 1070.6 mA/g, 939.9 mA/g, 755.9 mA/g, 462.6 mA/g, and 308.8 mA/g at a current density of 0.2, 0.5, 1, 2 and 3 A g−1, respectively. When the current density recovers to 0.2 A/g, the MoO3/C provides a discharge capacity of 938 mA/g. In comparison with pristine MoO3, it is clear that MoO3/C has a better rate performance; from the EIS results, it is acceptable that the improvement in the rate performance of MoO3/C is mainly due to the increase in conductivity resulted from the existence of amorphous carbon.
As we know, the available capacity of LIBs is strongly related to their operation temperature. Therefore, it is important to explore the electrochemical performance of MoO3/C at different temperatures. Fig. 5a shows the cycle performance of MoO3/C conducted at various temperatures. The cell with MoO3/C anode material was tested at 30 ℃ in the first 10 cycles and the last 10 cycles, and at 0 ℃, 20 ℃, 30 ℃, 40 ℃, 50 ℃, 60 ℃ from the 11th to the 100th cycle. The initial capacity difference is usually related to the initial condition of the electrode materials, such as the infiltration of electrolyte in active material, and the formation of SEI film. It can be seen from Fig. 5a that the lower the temperature is, the lower the specific capacity is, defining the temperature of 30 ℃ as the optimum value (11th-12th cycles). The relationship between discharge capacity (12th cycle) and test temperature is plotted in Fig. 6. The capacity retentions of MoO3/C are 75.2%, 67.6%, 66.3%, 38.9%, 17.0% and 6.8%, respectively, when measured from 0 ℃ to 60 ℃. The relative low retentions at higher temperatures may stem from the instability of the electrode, the decomposition of the electrolyte or both. It was concluded that LiPF6 can react with the trace amount of H2O (in ppm level) at elevated temperature, which results in the formation of some acids, e.g. PF5 and HF [37]. as a consequence, the active material of MoO3 will be corroded by these acids, leading to the decline of electrochemical performance. The corrosion of the active material at elevated temperature should be irreversible. This is the reason why the 101st to 110th capacity measured at 30 ℃ are obviously different, i.e. the capacity for 101st to 110th cycle measured after previous 100 cycles at relative low temperature (≤ 20 ℃) is significantly higher than those after previous 100 cycles at higher temperature (≥ 40 ℃).
Fig. 5.
(a) cycle performance, (b) CV curves, (c) EIS plots and equivalent circuit and (d) Z’ vs. ω−1/2 plots of MoO3/C at various temperatures.

To further understand the effect of operation temperature on the capacity, CV and EIS were carried out at various temperatures. The experimental results are presented in Fig. 5b and 5c. The redox peaks in CV curves measured at 0 ℃ are smaller than those at higher temperatures. This means the kinetics of redox reaction is restricted at low temperature. Furthermore, the redox peak current and the integrated area surrounded by CV curve increase gradually as increasing the test temperature, i.e. the effect of operation temperature on specific capacity is positive. As the arrows depicted in Fig. 5b, the oxidation potentials decrease and the reduction potentials increase with the temperature rise, indicating that the electrode polarization is reduced at relatively high temperatures. In fact, the polarization details can also be acquired by EIS spectra. The Nyquist plots (Fig. 5c) obtained at various temperatures, as well as their fitting results (Fig. 6), reveal that there is a temperature dependence on the polarization. Generally, the kinetics of the Faradic reaction becomes sluggish as the value of Rct increases [38]. Fig. 6 clearly shows that the sensitivity of Rct with respect to temperature is much stronger than that of Rs and Rf. And the value of Rct is also much larger than that of Rs and Rf. Therefore, the influence of resistance with the change of temperature in the cell equivalent circuit arises mainly from Rct
Furthermore, the Li ion diffusion rate is another key factor that influences the electrochemical performance of MoO3/C. The EIS result in low frequency region which is related to Warburg impedance (ZW) was employed to obtain the value of DLi. The value of ZW can be evaluated by following equation [39]:

where ω is the radial frequency, σw is the Warburg impedance coefficient and can be derived from linear fitting of Z’ vs. ω−1/2 plot (Fig. 5d). Then, DLi can be obtained:

where R is the gas constant (8.314 J·mol−1·K−1), T is the absolute temperature (in K), A is the surface area of the electrode, F is the Faraday constant (96,485 C/mol), σw is the Warburg impedance coefficient obtained from Eqn. (3). C ( C = ∫idt/FVm ) is the molar concentration of Li ion in the electrode and Vm is the molar volume of MoO3 (Vm = 30.7 cm3/mol). The plot of lnDLi vs. 1/T is exhibited in Fig. 7. It can be seen that the temperature dependence of DLi follows Arrhenius behavior. The modified Arrhenius equation for DLi is
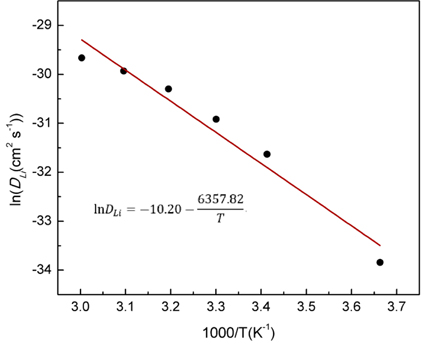

where D0 is the pre-exponential factor, Ea is the activation energy, R is the gas constant, T is the absolute temperature. Obviously, the relationship between lnDLi and 1/T is linear and the activation energy can be obtained from the slope of the fitting line. The value of Ea of MoO3/C is about 52.86 kJ/mol. It is confirmed that the electrochemical lithiation reaction rate for conversion-type anode materials is mainly controlled by the diffusion of Li ion [36]. From Fig. 7, it can be seen that the value of lnDLi decreases as decreasing temperature. Thus it can be concluded that the effect of temperature on the electrochemical lithiation reaction rate is positive. Therefore, it can be well understood that the polarization is aggravated by the reduction in DLi and the increase of Rct as decreasing temperature, corresponding to a poor electrochemical performance.
4. Conclusions
MoO3/C was prepared by using PANI doped with AMo as precursor. In this synthetic route, AMo was homogeneously dispersed in PANI resulting in an even carbon distribution in MoO3. Electrochemical measurements demonstrate that the electrochemical performance of MoO3/C at room temperature is significantly improved as compared with that of pristine MoO3. The superior electrochemical performance results from the abundant carbon in MoO3/C, which enhances the electrical conductivity of the electroactive material and buffers the expansion/contraction of MoO3 particles during the lithiation/delithiation process. The electrochemical measurement results indicate reduction in diffusion coefficient of lithium ions and increase of charge transfer resistance aggravate the polarization, that decreases the specific capacity as temperature is progressively decreased.




