


1. Introduction
Global warming, climate change, and fossil fuel exhausts have inspired the development of clean power sources. Furthermore, the rapid development of electric vehicles and energy storage systems has increased the demand for advanced and cheap lithium-ion batteries [1-7]. The development of high-performance cathode materials is an important research topic for enhancing the electrochemical properties of lithium-ion batteries [8-13]. Recently, manganese-based spinels (LiMn2O4) received attention because of their low cost and safety advantages [13-16]. However, there are still several issues that need to overcome, such as an insufficient rate capability due to low electronic conductivity and an unstable cyclic performance attributed to surface and structural instabilities [17-21]. Carbon coating is a possible approach to improve the electrochemical performance of LiMn2O4. A carbon surface layer with a high electronic conductivity can compensate for the low conductivity of LiMn2O4 and suppress the surface instability (e.g., Mn dissolution) [22-26]. Generally, an organic material decarbonization process is used to prepare a carbon layer on the surface of the cathode.
Here, a LiMn2O4 cathode was carbon-coated using a polydopamine layer as the carbon source. Polydopamine provides a strong adhesion and can activate secondary surface-mediated reactions [27-30]. Moreover, it can act as an organic precursor for N-doped carbon, which is beneficial for lithium ion transport at the interface [31-33]. The polydopamine film can be easily prepared through a spontaneous oxidative polymerization from dopamine (2-(3,4-dihydroxyphenyl)ethylamine) in a buffer solution (pH 8.5). The layer thickness can be controlled by the reaction time, which provides control of the carbon content. The surface carbon layer is expected to provide an enhanced rate capability to LiMn2O4 owing to the high electronic conductivity of carbon. Moreover, the carbon layer may reduce the unwanted side reactions between the reactive electrolyte containing HF, which can suppress Mn dissolution and enhance the cyclic performance of LiMn2O4.
2. Experimental Section
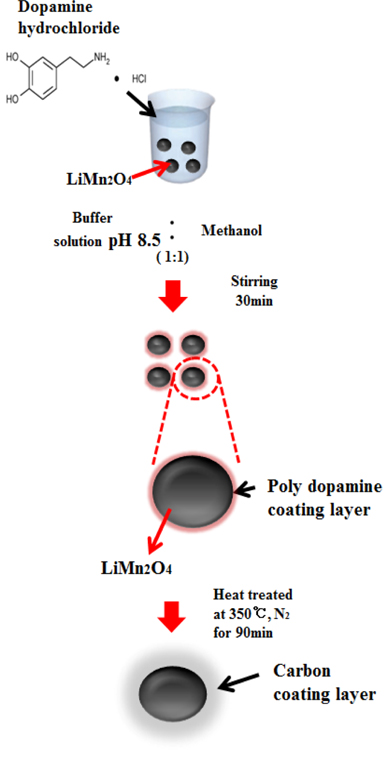
The LiMn2O4 nanoparticles were synthesized according to preexisting procedures [12,18]. The carbon coating was prepared using a polydopamine layer formed on the surface of the LiMn2O4 nanoparticles through a carbonization process during heattreatment. LiMn2O4 nanoparticles were immersed into a dopamine solution containing tris(hydroxymethyl)aminomethane (Tris) buffer solution (10 mM, pH 8.5, Aldrich) and methanol (99.9 %, Aldrich) cosolvents (CH3OH:buffer = 1:1 v/v). The solution was stirred at room temperature to suspend all nanoparticles in the mixed solution. A polydopamine layer formed on the surface of the LiMn2O4 nanoparticles through a polymerization process. Different stirring times (30 min and 90 min) were used to control the thickness of the polydopamine layer, which determined the carbon content after carbonization. For carbonization, the suspension was centrifuged, washed with ethanol, and heat-treated at 350 Ōäā for 90 min under a N2 atmosphere. The fabrication process for the carbon-coated LiMn2O4 nanoparticles is illustrated in Fig. 1.
Fig.┬Ā1.
Fabrication process for carbon-coated LiMn2O4 nanoparticles using a polydopamine layer as the N-doped carbon source.
The structure of each sample was analyzed by X-ray diffraction (XRD, Rigaku) using monochromatic Cu-K╬▒ radiation (╬╗ = 1.5406 ├ģ), and the sample morphologies were examined using a transmission electron microscope (TEM, JEOL-4010). The carbon content of the carbon-coated LiMn2O4 nanoparticles was determined using CHN elemental analysis (Thermo Fisher). For electrochemical testing, a cathode slurry was prepared by mixing LiMn2O4 (pristine and carbon-coated LiMn2O4), carbon black (Super P), and polyvinylidene fluoride in a weight ratio of 7:2:1, respectively. The mixed slurry was coated onto the surface of an Al foil using a doctor blade and subsequently dried at 90 Ōäā for 24 h. A coin-type cell (2032) was prepared using a cathode, a separator, a Li-metal anode, and an electrolyte (1 M LiPF6 in ethylene carbonate/dimethyl carbonate (1:1 v/v)). The cells were subjected to galvanostatic cycling (WonATech voltammetry system) over 4.3-3.5 V at various charge-discharge rates. Impedance measurements were performed by applying an AC voltage (5 mV amplitude, 100 kHz to 0.1 Hz frequency range) using an electrochemical workstation (AMETEK, VersaSTAT 3).
3. Results and Discussion
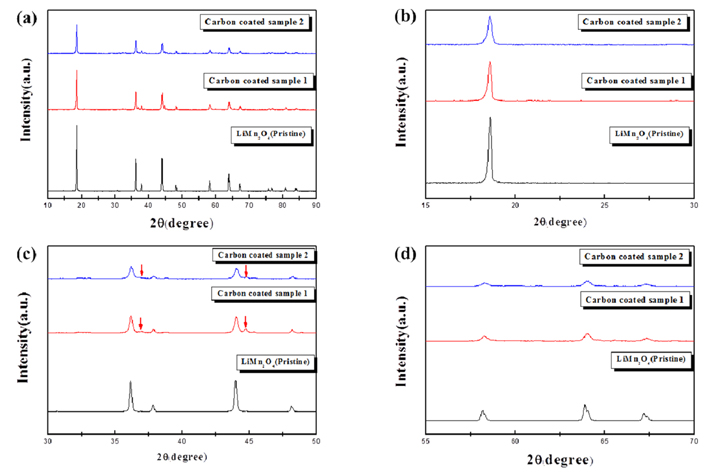
The surface morphologies of the pristine and carbon-coated LiMn2O4 were observed using TEM. The LiMn2O4 nanoparticles (diameter = 0.2-0.8 ┬Ąm) were much smaller than conventional cathode powders (generally over several ┬Ąm). The pristine LiMn2O4 had a smooth surface without any other heterogeneous particles (Fig. 2a). However, the surface of the carbon-coated LiMn2O4 particles appeared to be covered with a low-intensity film-like layer (Fig. 2b and 2c), which is likely the N-doped carbon derived from the carbonization of the polydopamine layer. The carbon content of the carbon-coated LiMn2O4 cathode was measured using CHN elemental analysis. The thickness of the polydopamine layer, which may determine the carbon content, was controlled by the polymerization time. The carbon content of the carbon-coated LiMn2O4 samples using the polydopamine layer was approximately 0.25 wt% and 0.65 wt% after 30 min and 90 min of reaction time, respectively. The former sample is hereafter referred to as ŌĆ£coated sample 1ŌĆØ and the latter as ŌĆ£coated sample 2ŌĆØ. Fig. 3a compares the XRD patterns of the pristine and coated samples. The three patterns corresponded to the typical spinel structure of LiMn2O4. However, the intensities of the diffraction peaks of the coated samples were lesser than that of the pristine samples. The pattern was examined in further detail by enlarging the peaks at angles of 15┬░-30┬░ (Fig. 3b), 30┬░-50┬░ (Fig. 3c), and 55┬░-70┬░ (Fig. 3d). The peaks of the coated sample were broadened and their intensity was reduced. Moreover, some unidentified peaks attributed to the 2nd phase were observed, as shown in Fig. 3c (marked with arrows). This indicates that the integrity of the LiMn2O4 structure slightly deteriorated, which may be associated with the oxygen loss during the carboncoating process. The carbon-coating layer was derived from the carbonization process of the organic material (polydopamine) through heat-treatment. A portion of oxygen in LiMn2O4 reacted with carbon during heating, which caused oxygen loss. However, the main frame of the spinel structure remained after the carbon-coating process, as shown in Fig. 3.
Fig.┬Ā2.
TEM images of (a) pristine LiMn2O4, (b) carbon-coated sample 1, and (c) carbon-coated sample 2.

Fig.┬Ā3.
XRD patterns of the pristine and carbon-coated samples. (a) Full range (10┬░-90┬░), (b) 15┬░-30┬░, (c) 30┬░-50┬░, and (d) 55┬░-70┬░.
The electrochemical properties of the pristine and carbon-coated LiMn2O4 were evaluated to determine the effect of preparing the carbon coating from a polydopamine layer. Fig. 4a presents the discharge capacities and cyclic performances of the samples measured at current densities of 70, 140, 280, 420, and 840 mA┬ĘgŌłÆ1 (voltage range of 4.3-3.5 V). pristine and coated sample 1 showed similar discharge capacities at low current densities (70 and 140 mA┬ĘgŌłÆ1, respectively). However, at high current densities, the discharge capacity of coated sample 1 was much higher than that of pristine sample, indicating that coated sample 1 had a superior rate capability than the pristine sample. The initial discharge capacity of coated sample 2 was lower than that for the other samples, which may be attributed to the oxygen loss (degradation of the phase integrity), as presented in the XRD analysis (Fig. 3). However, the conductive carbon layer enhanced the rate capability. Fig. 4b-d show the voltage profiles for the samples measured at current densities of 70, 280, and 840 mA┬ĘgŌłÆ1 (1st, 16th, and 36th cycles, respectively, in Fig. 4a). As shown in Fig. 4b, the discharge capacity of the pristine LiMn2O4 significantly decreased from ~115 to ~35 mAh┬ĘgŌłÆ1 as the current density increased from 70 to 840 mA┬ĘgŌłÆ1. The capacity retention of the pristine sample at 840 mA┬ĘgŌłÆ1 was only ~30% of that measured at a current density of 70 mA┬ĘgŌłÆ1. In contrast, the capacity fading depended on the current density (from 70 mA┬ĘgŌłÆ1 to 840 mA┬ĘgŌłÆ1) of coated sample 2 and was considerably lower than that of the pristine sample (from ~93 to ~68 mAh┬ĘgŌłÆ1). The capacity retention of coated sample 2 was ~73%. Table 1 summarizes the discharge capacities and capacity retentions of the samples measured at various current densities.
Fig.┬Ā4.
(a) Discharge capacities of pristine LiMn2O4, and the carbon-coated samples at current densities of 70, 140, 280, 420, and 840 mA┬ĘgŌłÆ1 in a voltage range of 4.3-3.5 V. Discharge profiles of the samples at 70, 280, and 840 mA┬ĘgŌłÆ1, (b) pristine LiMn2O4, (c) carbon-coated sample 1, and (d) carbon-coated sample 2.

Table┬Ā1.
Discharge capacities and capacity retentions of pristine LiMn2O4 and carbon-coated samples at various current densities. The percentages refer to the retention of the capacity at the 36th cycle compared with that at the 1st cycle.

The impedance spectra of the pristine and coated samples were measured prior to the electrochemical test to obtain more information about the sample electrodes. The Nyquist plots (Fig. 5) contain a broad semicircle and a straight sloping line. The broad semicircle may consist of two overlapped semicircles. Generally, a semicircle in a high-frequency range shows impedance attributed to a solid electrolyte interface, while a semicircle in a relatively low-frequency range represents the charge-transfer resistance [34,35]. The size of the semicircle depends on the impedance value. The semicircle sizes for the carbon-coated samples were considerably smaller than that for the pristine samples. Carbon-coated sample 2 had the lowest impedance among the three samples, which may explain its superior rate capability compared to the other samples (Fig. 4).
Fig.┬Ā5.
Nyquist plots of (a) pristine LiMn2O4, (b) carbon-coated sample 1, and (c) carbon-coated sample 2.
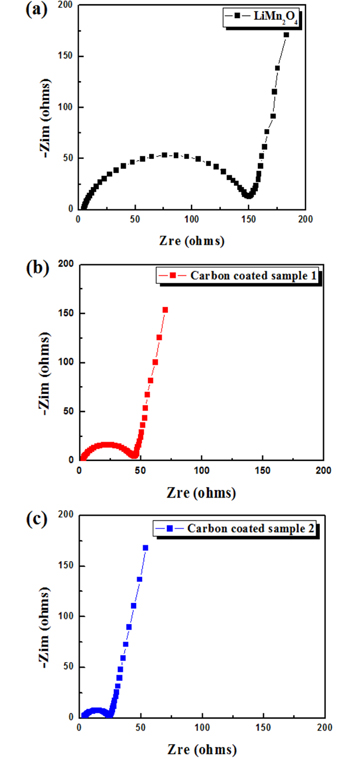
Fig. 6 presents the cyclic performance of the pristine and carbon-coated samples (current density = 140 mA┬ĘgŌłÆ1). The discharge capacity of the pristine LiMn2O4 gradually decreased during cycling because of the unwanted reaction with electrolytes (e.g., Mn dissolution). The small particle size of the LiMn2O4 particles may activate side reactions between the cathode and electrolyte because of their large surface area. However, the carbon-coated samples had an enhanced cyclic performance. Although the capacity of carbon-coated sample 2 was somewhat smaller, this stable cycle life may indicate that the carbon layer suppressed the surface reaction of LiMn2O4 with the electrolyte. Oxide, phosphate, and fluoride surface coatings can reduce side reactions with the electrolyte [36-41]. Although the carbon layer is a soft material, it also protects the surface of the cathode from reactive electrolytes in the same manner as hard coating materials. This protection may provide the enhanced cyclic performance shown in Fig. 6.

4. Conclusions
LiMn2O4 nanoparticles were carbon-coated to enhance the electrochemical performance of lithium-ion batteries. A polydopamine layer was introduced as the N-doped carbon source for carbon-coated LiMn2O4. The carbon was homogeneously coated on the surface of the LiMn2O4 nanoparticles; defects in the carbon are beneficial for lithium ion transport in the interface. However, the phase integrity deteriorated during the carbon-coating process due to oxygen loss from the LiMn2O4 structure. The rate capability and cyclic performance of the carbon-coated samples were superior to those of the pristine samples. This indicates that the carbon coating can act as a protective layer from the reactive electrolyte during cycling and as a conducting material. Thus, the LiMn2O4 nanoparticles represent a promising cathode material for use in lithium-ion batteries.




