1. Introduction
The application and storage of renewable energy is needed to cope with unpredictable climate changes and depletion of natural resources. To this end, development of power generation devices that are immune to environmental hazards have been actively investigated [1]. Advancement of power storage technologies, such as Li-ion batteries (LIBs), redox flow batteries, and metal-air batteries, have demonstrated noticeable progress. Among them, LIBs have evolved from portable electronics to electric vehicles (EVs) and energy storage systems (ESSs). The increasing concerns on climate change has led to stricter stipulations for electro-mobility, which demand rechargeable batteries with higher energy densities [2]. Advanced cathode materials are essential to develop LIBs with higher performances. As a result, the battery industry has witnessed various cathode materials over the past 30 years, even though only a few of them were successfully commercialized. Voltage and capacity fading, structural and thermodynamic instability, and poor ionic conductivity have been identified as the barriers for the development of advanced cathode materials [3,4]. On the other hand, lithium cobalt oxide (LiCoO2, LCO), first identified as a cathode material by Goodenough et al. [5], exhibits a high theoretical capacity and moderate Li+/electron conductivity, which are beneficial for a reasonable electrochemical performance [6ŌĆō8]. In addition, the legendary LiCoO2 or LCO is still actively utilized as an active cathode material in LIBs owing to its highly reproducible synthesis and superior electrochemical performance [9,10]. For this reason, it might be meaningful to revisit the LCO cathodes for a comprehensive exploitation of their theoretical electrochemical performance.
Even though the theoretical capacity of LCO is as high as 274 mAh gŌłÆ1, only up to 60% of this capacity has been practically utilized, which is approximately 165 mAh gŌłÆ1 [11]. When compared to Li/Li+, LiCoO2 experiences order-disorder phase transition, structural instability, and severe capacity loss when it is operated at more than 4.3 V. Charging up to 4.2 V results in an orderŌĆōdisorder transition from hexagonal to monoclinic structure, as approximately half of the Li+ ions are dislodged from the host structure. Upon further charging above 4.5 V, another transition occurs from O3 to H1ŌĆō3 or O6 phases, caused by various physio-chemical interactions inside the cathode [12ŌĆō15]. The orderŌĆōdisorder transitions extensively lower the Li+ diffusivity, and the transition to the H1ŌĆō3 phase critically destroys the structure of the cathode material despite the completely reversible phase transitions. Therefore, it is very essential to enhance the LCO cathodes in terms of the cutoff voltage and fast chargeability [16,17]. To this end, doping LCO with various metal ions has resulted in various degrees of success [18,19]. The incorporation of divalent ions into the LiO2-layer can stabilize the structure [20]; moreover, such divalent dopants can prevent cation mixing by compensating for the deficiency of Li+ ions in layered materials [21,22]. Among the divalent dopants, Ca2+ has been proposed as one of the effective dopants for improved cycling stability. Various methods have been developed for ion doping, such as co-precipitation, solid-state, solŌĆōgel, molten salt, and hydrothermal methods. Among these, solid state and co-precipitation methods have been actively utilized owing to their low cost and scalability.
In theory, a small particle size is beneficial to enhance the rate capability of cathode materials [23,24]. The reduced particle size shortens the diffusion pathways for Li+ ions, thus creating a large contact area between the active materials and the conductive additives [25]. The rate determining step of the discharging process at a high rate operation contributes to the solid-state diffusion of Li+ ions inside the electrodes [26,27]. The beneficial effects of reduced particle size include structural durability as a result of the lower concentration gradient developed inside the particles. High-energy ball milling has been a reliable process to reduce the particle size of cathode materials to sub-micron scales [28,29]. On the other hand, it has been reported that excessive nano-sizing is detrimental to the electrochemical performance of cathode materials [30,31], because it promotes side reactions at high voltages and leads to structural damage. Therefore, it is important to investigate the effect of particle size on the electrochemical performance of cathode materials that are prepared by various synthetic routes.
Herein, we report a comparative study on the electrochemical performance of Ca-doped LCO (LiCo1ŌłÆx CaxO2, x = 0.02) that were synthesized by two different routes, namely solid-state synthesis and co-precipitation. The solid-state synthesis of Ca-doped LCO resulted in a smaller particle size and better homogeneity of the dopants compared with the co-precipitated sample. This would result in a higher cycling stability and rate capability. Electrochemical simulations using various particle sizes supported the effective role of reduced particle size on the rate capability. These results have demonstrated the pivotal importance of reducing the particle size and homogeneous doping to improve the electrochemical performance of layered cathode materials, especially at higher operational voltages.
2. Experimental
2.1 Solid-state synthesis of Ca-doped LCO
For the solid-state synthesis of Ca-doped LCO, stoichiometric amounts of lithium carbonate (Li2-CO3; Junsei Chemical Co., Japan; 99%), cobalt oxide black (Co3O4; Junsei Chemical Co., Japan; > 85%), and calcium nitrate tetrahydrate (Ca(NO3)2┬Ę4H2O; Kanto Chemicals Co., Japan; 98.5%) were ball milled, calcined, and sintered. Essentially, 4.02 g of Li2CO3, 9.49 g of Co3O4, and 0.492 g of Ca(NO3)2┬Ę4H2O were initially mixed in an agate mortar and pestle, then thoroughly mixed and ground in a planetary ball mill (PM 100, Retsch Co., Germany) at 3000 rpm for 4 h. The mechano-chemically homogenized precursors were then calcined at 800┬░C for 12 h and cooled down naturally. Thus, the initially calcined powder was ground and mixed well before final sintering at 850┬░C for 24 h. All the calcination and sintering were carried out in an air atmosphere. The resultant material was labeled as SS-Ca-LCO, where SS denotes solid-state synthesis. For comparison, pristine LCO without doping was synthesized as well by solid-state route and labeled as SS-PLCO.
2.2 Co-precipitation of Ca-doped LCO
For comparison, a precursor for Ca-doped LCO was synthesized by co-precipitation of Co0.98 Ca0.02(OH)2. Aqueous solution of cobalt nitrate hexahydrate (Co(NO3)2┬Ę6H2O; Junsei Chemicals Co., Japan; 98.0%) and Ca(NO3)2┬Ę4H2O was prepared according to stoichiometry. Aqueous ammonia (NH3┬ĘH2O; Daejung Chemicals & Metals Co., Korea; 25ŌĆō28%) was added to the solution of metal salts, so that the concentrations of the metal ions and NH3 were 2.0 and 1.0 M, respectively, and the total volume of the solution was 100 mL. Separately, 1 M NH3 aqueous solution of 1 L was prepared and filled in the reactor beforehand. Subsequently, the metal and NH3 solution was added dropwise using a peristaltic pump (Multi-channel Pump, JiPump Co.; China) at a speed of 0.04 L hŌłÆ1 under an inert atmosphere of N2. Here, ammonia was employed as an auxiliary complexing agent, so that the precipitation of metal ions are retarded for more homogeneous composition of the particles [32]. During the co-precipitation reaction, the solution was vigorously agitated using an overhead stirrer at 350 rpm, with the temperature set to 60┬░C using a heating mantle. Simultaneously, a 4 M sodium hydroxide (NaOH; Yakuri Pure Chemicals Co., 96.0%) solution was added dropwise using a computerized peristaltic pump (Masterflex L/S computer-compatible drive, 07551-30 series, Cole-Parmer Co; USA) to maintain the pH value at 10.50 during the co-precipitation reaction. After 1 h of co-precipitation, the reactor was cooled down to ambient temperature and the precipitated solution was centrifuged for 10 min at 3000 rpm. Subsequently, the supernatant was carefully discarded and the precipitate was mixed with fresh deionized water. The centrifuging was repeated two more times. The precipitate was rinsed with deionized water several times and filtered before being dried for 2 h at 80┬░C in a vacuum oven. The resultant light pinkish-brown powder of Co0.98Ca0.02(OH)2 was mixed with a stoichiometric amount of lithium hydroxide monohydrate (LiOH┬ĘH2O; Junsei Chemicals Co., 98%) using an agate mortar and a pestle. The mixed powder was heated in a box furnace for 12 h at 800┬░C, at a heating rate of 100┬░C/hŌłÆ1 and a natural cooling rate. The calcined powder was ground and mixed well before finally being sintered at 850┬░C for 24 h. The Ca-doped lithium cobalt oxide (LiCo0.98Ca0.02O2) powder thus obtained, was ground in an agate mortar before the characterization. The resultant material was labeled as CP-Ca-LCO, where CP denotes co-precipitation.
2.3. Materials Characterization
For structural analysis, X-ray diffractometry (XRD; MiniFlex 600, Rigaku Co., Japan) was performed in a range of 10ŌĆō80┬░ (2╬Ė) with a step width of 0.02┬░ and scanning speed of 10┬░ minŌłÆ1. The particles were imaged using a scanning electron microscope (SEM; VEGA3, Tescan Co. and S3500, Hitachi Co.: Japan) and a scanning transmission electron microscope (STEM; Talos F200X, Thermo Fisher Scientific Co.; USA) The homogeneity of dopants in Cadoped LCO particles and other elemental information was obtained using energy dispersive spectroscopes (EDS) that were attached to the SEM and TEM instruments.
2.4. Electrochemical Characterization
The electrodes were prepared by coating a slurry of N-methyl-2-pyrrolidone (NMP; Sigma Aldrich, USA; 99.5%) that is mixed with Ca-doped LCO, conductive carbon black (Super-P, Timcal Co.), and polyvinylidene fluoride (PVDF; KF1100, Kureha Co.) in a mass ratio of 6:2:2 on an aluminum foil of 20 mm using a doctor blade. The coated electrode was dried at 80┬░C under vacuum for 2 h. Next, the dried electrode was roll-pressed and punched into discs of 0.95 cm2 with an active material loading of 1ŌĆō2 mg. Coin cells of 2032-type were assembled with the coated electrode as the cathode and a lithium foil as the anode, both of which sandwiched a separator that was soaked with an electrolyte (Panax-Etec Co.; Korea) of 1M lithiumhexafluorophosphate (LiPF6) in ethylene carbonate and ethyl-methyl-carbonate (EC:EMC, 3:7 w/w) inside a glove box (Moisture Oxygen Technology (MOTEK) Co.; Korea), where the moisture and oxygen levels were kept below 1 ppm.
2.5. Electrochemical Simulation
A one-dimensional single particle model (SPM) was assumed to simulate a battery system that includes the LiCoO2 cathode, the electrolyte, and the Li metal anode. The open circuit potential was determined as a function of intercalation quantity (i.e., x in LixCoO2) by galvanostatic intermittent titration technique (GITT). The electrochemical (de)intercalation at the electrode-electrolyte interface was described by FickŌĆÖs first law and the Butler-Volmer equation; and the solid-state chemical diffusion of Li-ions in the electrode particle was modeled by FickŌĆÖs second law. Exchange current density for the (de)intercalation of Li+ ions was set to 0.5 mA cmŌłÆ2 for all the simulations in this work. Conductivity, transference number, and thickness of the electrolyte were set to 10 mS cmŌłÆ1, 0.3, and 20 ╬╝m, respectively, which are close to the specification of commercial electrolytes for LIBs. The conductivity of the electrolyte was assumed to be a function of concentration according to KohlrauschŌĆÖs law. Using this SPM, batteriesŌĆÖ electrochemical performances were simulated with respect to the LCOŌĆÖs particle size, C-rate, and solid-state diffusivity. The time-dependent partial differential equations in weak formulations were numerically solved by finite element method (FEM) using COMSOL Multiphysics 5.5 software at Precision 7920 workstation (DELL Co.) with 24 cores.
3. Results and Discussion
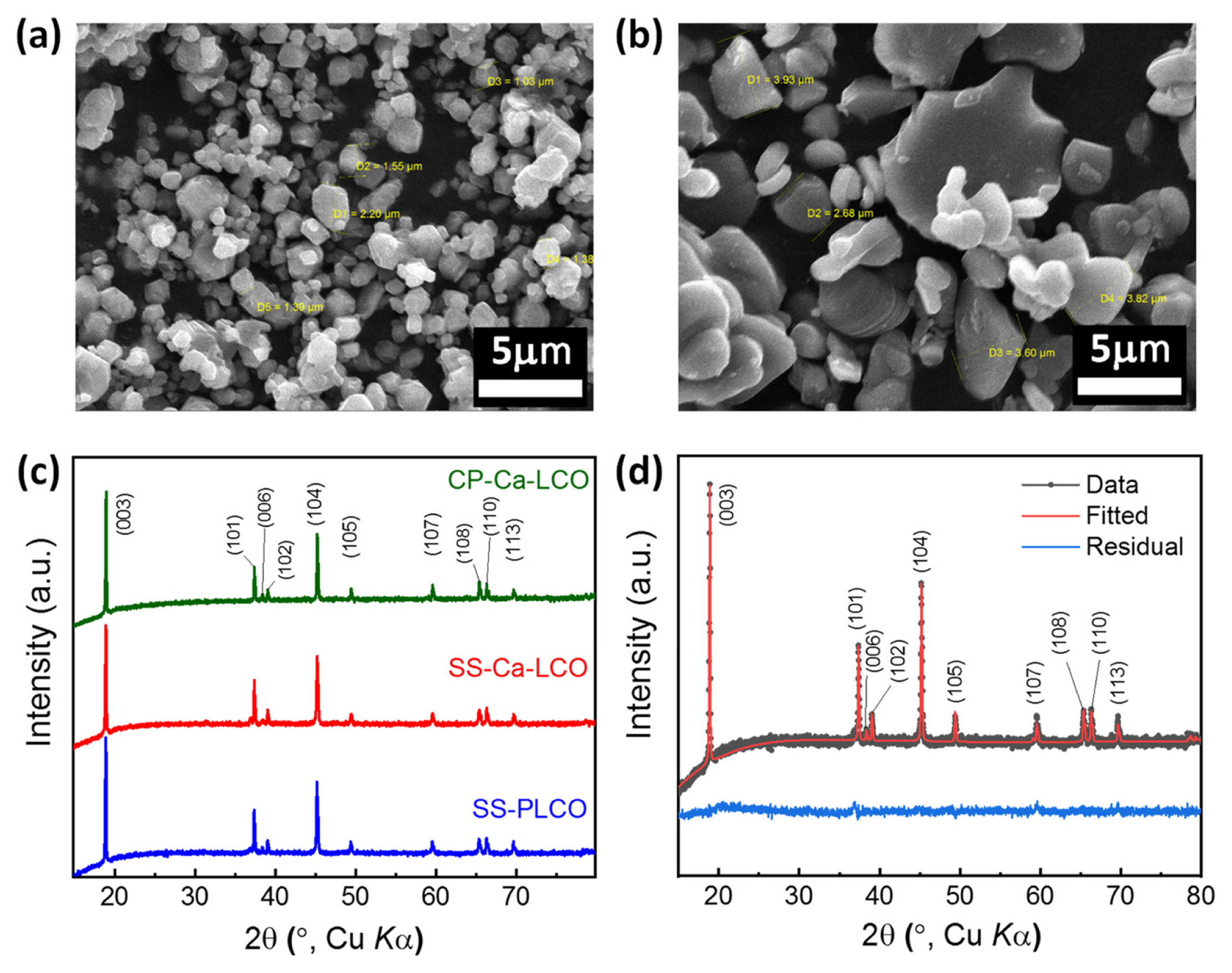
Figs. 1(a) and 1(b) show the SEM images of LiCo0.98Ca0.02O2 powders that were synthesized by solid-state (SS-Ca-LCO) and co-precipitation (CP-Ca-LCO) routes, respectively. SS-Ca-LCO exhibited smaller and more uniform particle size compared to CP-Ca-LCO. The morphology particle size of SS-PLCO were similar to that of SS-Ca-LCO (Fig. S1). The average particle sizes (i.e., diameter, d) were approximately 1.5 and 3.8 ╬╝m for SS-Ca-LCO and CP-Ca-LCO, respectively. This signifies that the mechano-chemical route (e.g., high-energy ball milling) could provide a smaller and more evenly distributed particle size distribution with less agglomeration compared to wet-chemical routes, such as co-precipitation. The nucleation and growth of the coprecipitated precursors are determined by various factors such as feeding rate, concentration of solutions, stirring rate, and reactor volume [33]. We speculate that the growth of the precursors were particularly preferred by the experimental condition of coprecipitation in this work. Another possible factor for the larger particle size of CP-Ca-LCO is the manual grinding with mortar and pestle. Fig. 1(c) shows the XRD patterns of Ca-doped LCO from the two different synthetic routes compared with that of pristine LCO from solid-state synthesis. The diffraction patterns of Ca-doped LCO matched with the pattern of pure LiCoO2, which was a hexagonal structure of layered sodium ferrite (╬▒-NaFeO2) with the space group of RŌłÆ3m. The structural information was further determined using Rietveld refinement, as shown in Fig. 1(d) and Table 1. The c/a values of the all the prepared samples were found to be more than 4.99, which were higher than the critical value of 4.9; any value lower than the critical value signifies the development of a disorderly rock salt structure. I(003)/I(104), the ratio of intensity for (003) plane to that for (104) plane, has been utilized as a useful criteria to determine the degree of ŌĆ£cation mixing,ŌĆØ which is the exchange of Li+ and transition metal ions that is detrimental to the electrochemical performances [34]. I(003)/I(104) ratio that is lower than 1.2 indicates a significant degree of cation exchange. The I(003)/I(104) ratio was larger than 1.2 for all the samples, indicating that cation mixing did not occur.
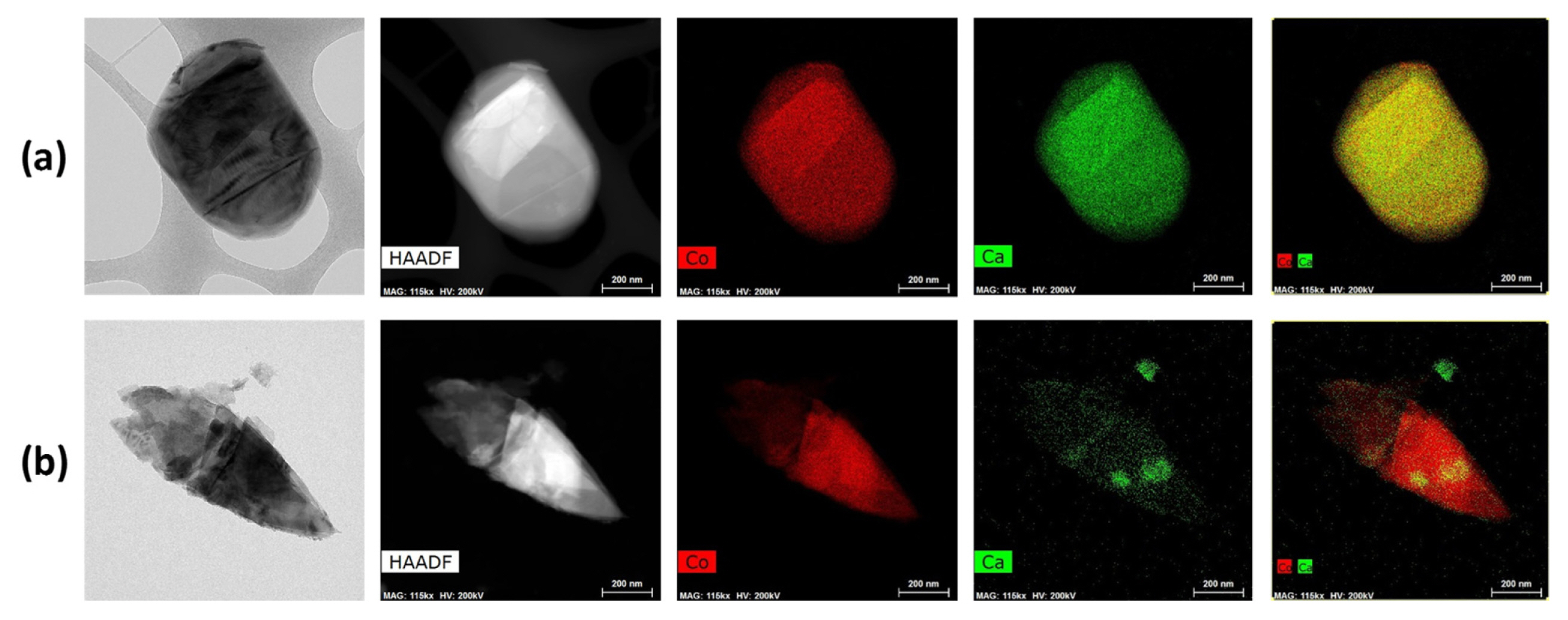
Fig. 2 shows the STEM images in bright and dark-field modes together with elemental maps for Ca-doped LCO. Among them, SS-Ca-LCO represented more homogeneous distribution of Ca compared to CP-Ca-LCO. The less homogenous distribution of Ca2+ ions in the CP-Ca-LCO sample may be deeply related with the non-homogenous coprecipitaiton of Ca2+ ions in the hydroxide precursor. The solubility product (Ksp) of Ca(OH)2 is 6.5├Ś10ŌłÆ6, which is significantly larger than 1.3├Ś10ŌłÆ15 for Co(OH)2; this means that the precipitation of Co(OH)2 can be kinetically much more preferred compared to Ca(OH)2. The different kinetics of precipitation for Co2+ and Ca2+ may have led to the non-homogeneous agglomeration. For such cases, solid-state mechanochemical doping can be more beneficial for homogeneous distribution of dopants. EDS analysis provided evidence that the proportion of calcium (x) in LiCo1ŌłÆxCaxO2 was approximately 0.019 and 0.017 for SS-Ca-LCO and CP-Ca-LCO, respectively (Fig. S2). These values were close to the targeted value of 0.02.
Fig. 3(a) shows the electrochemical charge/discharge profiles of SS-PLCO, SS-Ca-LCO, and CP-Ca-LCO in the potential range of 3.0 and 4.5 V vs Li/Li+ at 0.1C rate (i.e., 1C = 274.89 mA gŌłÆ1, corresponding to the theoretical capacity of LCO). The initial capacity was 180 mAh gŌłÆ1 and 169 mAh gŌłÆ1 for Ca-doped LCOs and pristine LCO, respectively, evidencing the enhanced electrochemical performance of Ca-doped LCOs. Profiles of both Ca-doped LCOs coincided significantly with each other, with an initial discharge capacity of approximately 180 mAh gŌłÆ1. This signifies that both the samples shared similar material properties as electrode materials. The cycle test at 0.1C in the range of 3.0ŌĆō4.5V vs Li/Li+ yielded a higher capacity retention of 94.2% for SS-Ca-LCO compared to 91.1% for CP-Ca-LCO after 25 cycles. On the other hand, SS-PLCO exhibited continually lower capacity compared to the Ca-doped LCOs throughout the cycling. Moreover, the long-term cycling of 100 cycles at accelerated rate of 0.5C exhibited higher cycleability of Ca-doped LCO compared to pristine LCO (Fig. S3), evidencing the enhanced structural stability by Ca-doping. SS-Ca-LCO also exhibited better rate capability compared to CP-Ca-LCO as shown in Figs. 3(c) and 3(d). The specific capacity at 5C (i.e., charging or discharging batteries in 12 min) was 91.95 and 69.64 mAhgŌłÆ1 for SS-Ca-LCO and CP-Ca-LCO, respectively; these values correspond to 62.9% and 47.2% retention with respect to the initial capacity values.
The better cycle and rate performances of SS-Ca-LCO compared to CP-Ca-LCO could be ascribed to the following factors: (1) smaller particle size and (2) more homogeneous distribution of Ca2+ dopants. Smaller particle size enabled a decrease in the pathway for ionic diffusion (l), thus reducing the time constant for ionic diffusion according the relationship t ~ l2/D; here the solid-state chemical diffusion coefficient (D) is a constant. The reduced time constant signifies a faster diffusion in the cathode materials, which would enable a higher rate capability. Moreover, a faster transport of ions in smaller particle size resulted in a smaller concentration gradient (dc/dr); here c denotes the Li+ concentration as a function of the radial distance (r) from the center of a particle. As the concentration gradient might result in a mechanical stress, a smaller particle size could minimize the mechanical stress and provide a better cycle performance. On the other hand, a better homogeneity of Ca2+ ions within LCO layers could strengthen the structure against the insertion/extraction processes during the batteriesŌĆÖ operation. These manifold effects may have resulted in a better rate and cycle performances of SS-Ca-LCO compared to those of CP-Ca-LCO.
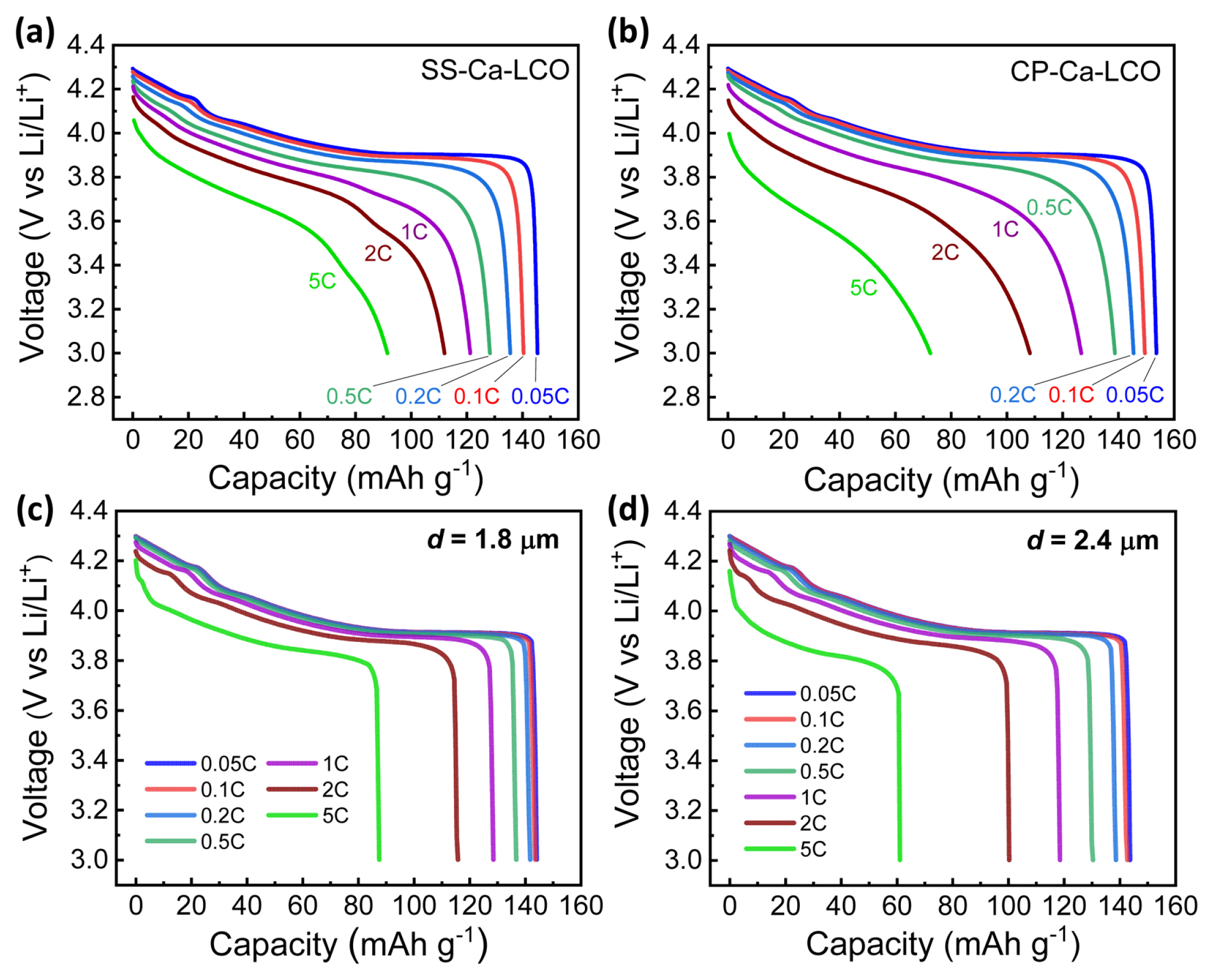
The discharge voltage profiles of SS-Ca-LCO and CP-Ca-LCO at different C-rates are shown in Figs. 4(a) and 4(b), respectively. Not only the capacity but also the average voltage decreased with the C-rate, implying a more significant reduction in the delivered energy. It was clearly observed that SS-Ca-LCO with the smaller particle size exhibited higher average voltage and specific capacity compared to CP-Ca-LCO, which had a larger particle size. The capacity was mainly determined by the time constant for the diffusion of Li+ into the center of the particles. As was discussed in the previous paragraph, SS-Ca-LCO with a smaller time constant could exhibit a larger capacity at high rates compared to CP-Ca-LCO. On the other hand, the average voltage was reduced by ohmic, charge transfer, and concentration polarization. Among them, the ohmic polarization could be approximated to be constant because it was mainly owing to the electrolyte resistance, being a function of the separatorŌĆÖs thickness. Smaller particle size contributed to reducing the overpotential in the following two mechanisms: (1) the increase in the specific surface area reduced the current density and hence, the charge transfer overpotential; (2) the decrease in the length of solid-state Li+ diffusion reduced the concentration overpotential.
To investigate the influence of the particle size on the electrochemical performance, a numerical simulation was conducted using a single particle model (SPM) of LCO at 0.05C, 0.1C, 0.2C, 0.5C, 1C, 2C, and 5C (Fig. S4). Figs. 4(c) and 4(d) simulate the voltage profiles for particle sizes (i.e., diameter) of 1.8 and 2.4 ╬╝m, respectively. The particle sizes were selected to match the simulated voltage profiles with the experimental data. The experimental particle diameter was 1.5 and 3.8 ╬╝m for SS-Ca-LCO and CP-Ca-LCO, respectively; and the discrepancies in the diameter may be due to the uncertainties in determining the average particle size of CP-Ca-LCO with the broad distribution of the particle size. Nevertheless, the SPM simulation provides useful understanding on the rate capability by different particle size. These observations clearly reveal that the rate capability was sensitive to the particle size of the cathode materials. The significant differences between the average voltage for the simulated and experimental profiles could be attributed to the fact that the experimental data was measured for the composite electrodes, whereas the simulation assumed single particle for clarity and simplicity.
4. Conclusions
Average particle size and homogeneity of the dopant closely depended on the synthetic routes of Ca-doped LiCoO2 cathode materials. Compared with the co-precipitation, the solid-state synthesis resulted in a smaller particle size and better homogeneity of Ca2+ dopant, which positively influenced the rate and cycle performance. The better cycle performance of Ca-doped LiCoO2 from solid-state synthesis was attributed to the enhanced structural stability; which was owing to an effective doping and less severe structural damage in accordance with the alleviated concentration gradient in the smaller particles. The reduced particle size effectively decreased the length for solid-state diffusion, which enabled better rate capability compared to the Ca-doped LiCoO2 from co-precipitation. The positive role of the particle size reduction in enhancing the rate capability was also evidenced by electrochemical simulations based on a single particle model of LiCoO2 cathodes. Thus, this work established the intertwined effects of particle size and doping on the electrochemical performance of cathode materials for Li-ion batteries.