1. Introduction
Integration of intermittent renewable energy resources (solar and wind) into grids require utilization of efficient energy storage technologies. Higher specific powers and longer cycle lives of supercapacitors make them particularly appropriate for grid applications as they can buffer short-term power fluctuations [1,2]. However, supercapacitors exhibit significantly lower specific energy than batteries. Thus, low-cost electrode materials with simultaneously high specific power and specific energy must be engineered by scalable methods to exploit the full potential of supercapacitors and to push the technology forward [3,4].
Pseudocapacitive electrode materials (transition metal oxides, conductive polymers) store energy via reversible charge transfer (faradaic) reactions at the electrode surface. They offer great potential to overcome specific energy limitation as they can achieve higher capacitances compared to carbonaceous materials (graphene, activated carbon, CNT, etc.) that store energy statically at the electric double layer (EDL) [4]. Manganese dioxide (MnO2) was first exploited as a pseudocapacitive material by Lee and Goodenough in 1999 [5]. The intense interest in MnO2 for supercapacitor applications is driven by its low toxicity, high theoretical capacitance (1370 F gŌłÆ1 over a potential window of 0.8 V, assuming the transfer of one electron per Mn atom) [6], and low cost. MnO2 exhibits a rich polymorphism as its building [MnO6] octahedral units form numerous three-dimensional structures with tunnels or interlayers that can facilitate ion intercalation. The birnessite-type manganese dioxide (╬┤-MnO2) consists of stacked monolayers (0.7 nm interlayer distance) of edge-shared [MnO6] octahedra and has the most open structure of all MnO2 polymorphs [7]. However, low electrical conductivity (10ŌłÆ5ŌĆō10ŌłÆ6 S cmŌłÆ1) and poor cycling stability hinder electrochemical performance of MnO2 [5]. To overcome these disadvantages, MnO2 has been incorporated by carbonaceous materials, graphene and its derivatives or conductive polymers [8ŌĆō11].
Besides being used as a conductive additive, graphene can function as a sacrificial template in synthesis of MnO2 nanostructures with controlled morphology and improved electrochemical performance. Graphene oxide (GO) [12] and reduced graphene oxide (rGO) [13] were reported to induce the growth of ╬┤-type MnO2 by in-situ replacement of graphene carbons by edge-sharing [MnO6] octahedra when used as sacrificial templates. However, the synthesis of GO/rGO is a laborious multi-step process which requires the use of harsh chemicals [9]. Moreover, dense oxygen groups and defects on GO and rGO hinder homogeneous distribution of MnO2 particles and lead to irregular morphologies, whereas ŽĆ-conjugated defect-free graphene enables efficient templating. Lately, an environmentally benign single-step liquid-phase exfoliation (LPE) has become a prominent non-oxidative method in bulk production of defect-free graphene [14,15]. Chen et al. utilized graphene, exfoliated in an organic solvent N-methyl-pyrrolidone, as a sacrificial template in synthesis of MnO2 nanolamellas and reported a specific capacitance of 206 F gŌłÆ1 [16].
In this work, a defect-free graphene was obtained by a scalable and environmentally benign surfactant-assisted liquid-phase exfoliation (LPE) in water. To our knowledge, this is the first report on the use of water-driven exfoliated graphene as a sacrificial template in synthesis of ╬┤-MnO2 under hydrothermal conditions. Graphene-templated MnO2 exhibited a specific capacitance higher than that of its previously reported carbon and conductive polymer composites. PEDOT: PSS coating further enhanced the capacitance and the rate capability of the MnO2. Asymmetric supercapacitor composed of MnO2/PEDOT:PSS (positive electrode) and Fe3O4/C (negative electrode) promises for future practical applications with its high specific energy (18 Wh kgŌłÆ1), specific power (4.5 kW kgŌłÆ1) and cycling stability (85% capacitance retention after 1000 cycles).
2. Experimental
2.1 Preparation of colloidal graphene
The cationic surfactant-stabilized graphene dispersions were prepared by sonicating a mixture of 500 mg of graphite powder (< 20 ╬╝m, Sigma Aldrich) and 25 mg of hexadecyl trimethyl ammonium bromide (CTAB, Sigma Aldrich) in deionized (DI) water (50 mL) with a Bandelin Sonopuls HD3200 ultrasonic homogenizer (microtip MS 73, dia. 3 mm). The sonicator was operated at 65% amplitude of 200 W and 20 kHz for 1.5 h in the pulse mode (1-s on, 0.5-s off) in an ice bath. The resulting dispersion was left to sit for ~20 h allowing aggregation to form and then centrifuged (Allegra X-30R, Beckman Coulter, USA) at 2000 rpm for 30 min. The precipitate was discarded and supernate was collected for further use. The yield was recorded as ~0.4 mg mlŌłÆ1.
2.2 Preparation of exfoliated graphene-induced MnO2
315 mg potassium permanganate (KMnO4, Kimetsan) was mixed with 30 mL of exfoliated graphene dispersion (~0.4 mg mLŌłÆ1) and hydrothermally treated for 8 h at 160┬░C in a stainless steel autoclave. Before drying at 80┬░C, the sample was washed with ethanol and DI water.
2.3 Preparation of MnO2/PEDOT:PSS nanocomposite
200 mg of MnO2 powder was dispersed in 30 mL DI water, then 0.5 mL of PEDOT:PSS solution (solid content: 3.0 ŌĆō 4.0 wt% in H2O, Sigma Aldrich) was added into the dispersion and vigorously stirred overnight. The product was washed with ethanol and DI water and dried at 60┬░C under vacuum.
2.4 Material Characterizations
QUANTA 400F field emission scanning electron microscope (FE-SEM) and JEOL JEM-2100F transmission electron microscope (TEM) were used to investigate structural properties of samples. Before SEM imaging, samples were coated with a thin Au-Pd layer (2ŌĆō5 nm). EDS (Energy dispersive X-ray spectroscopy) mapping was performed by ZEISS/GeminiSEM 300 with a Bruker X-Flash 100 detector. Rigaku Ultima-IV diffractometer equipped with a CuK╬▒ source was used to perform X-Ray diffraction (XRD) studies (scan rate: 0.6┬░ minŌłÆ1). Raman spectra were recorded with 532 nm laser excitation (Renishaw InVia dispersive Raman microscope). X-ray photoelectron spectroscopy (XPS) analyses were performed on a PHI 5000 VersaProbe instrument with a monochromatic AlK╬▒ source. Samples were evacuated at 200┬░C for 10 hours and subjected to nitrogen (77 K) gas adsorption-desorption using a Quantachrome Autosorb-6 instrument. Brunauer-Emmett-Teller (BET) method and Density Functional Theory (DFT) were used to determine the specific surface area and pore size distribution, respectively.
2.5 Electrochemical measurements
Active material, polyvinylidene fluoride binder (PVDF, Kynar® HSV 900) and conductive carbon black (Timcal Graphite & Carbon Super P®) were mixed in N-methylpyrrolidone (NMP, Merck) at 80:10:10 weight ratio. Then, this slurry was coated onto a stainless steel foil (MTI) in the form of a 500 μm thick film. After drying overnight at room temperature, the electrodes were further dried under vacuum (60°C, 4 h).
For the three-electrode measurements, a platinum wire and a silver/silver chloride (Ag/AgCl sat. with KCl (aq)) electrodes served as the counter and reference electrodes, respectively. The mass loading of the working electrodes was ~3 mg cmŌłÆ2. Cyclic voltammetry (CV) and galvanostatic charge-discharge (GCD) tests were realized in 1 M sodium sulphate (Na2SO4, Sigma Aldrich) aqueous solution (0 ŌĆō 1.2 V vs. Ag/AgCl) by using a Gamry potentiostat/galvanostat (Reference 3000). The specific capacitances Cs (F gŌłÆ1) were calculated with respect to the following equation: Cs = (I x ╬öt)/(m x ╬öV) where ╬öV (V) is the width of potential (the IR-drop excluded), I (A) is the discharge current, m (g) is the mass of active material and ╬öt (s) is the discharge time
2.6 Fabrication of asymmetric supercapacitors
To assemble an asymmetric supercapacitor (ASC), a glass fiber separator (Millipore┬«, thickness: 475 ╬╝m) saturated by 1M Na2SO4 aqueous electrolyte was sandwiched between the negative Fe3O4/Carbon (reported in our previous work [17]) and positive MnO2 or MnO2/PEDOT:PSS electrodes. The charge balance (q+=qŌłÆ) was ensured by adjusting the mass ratio of positive/negative electrodes (q= Cs x ╬öV x m). CV, GCD, and cycle life measurements were performed within a potential range of 0 to 2.0 V. The following equations were used to calculate specific energy Ecell (Wh kgŌłÆ1) and specific power Pcell (W kgŌłÆ1) of the cell: Ecell = (0.5 x Cs,cell x╬öV2)/3.6 and Pcell= E/t x 3600 where Cs,cell is the specific capacitance of the cell and t (s) is the discharge time. The Coulombic efficiency was calculated according to: ╬Ę = (td/tc) x 100 where tc and td refer to total charge and discharge times, respectively.
3. Results and Discussion
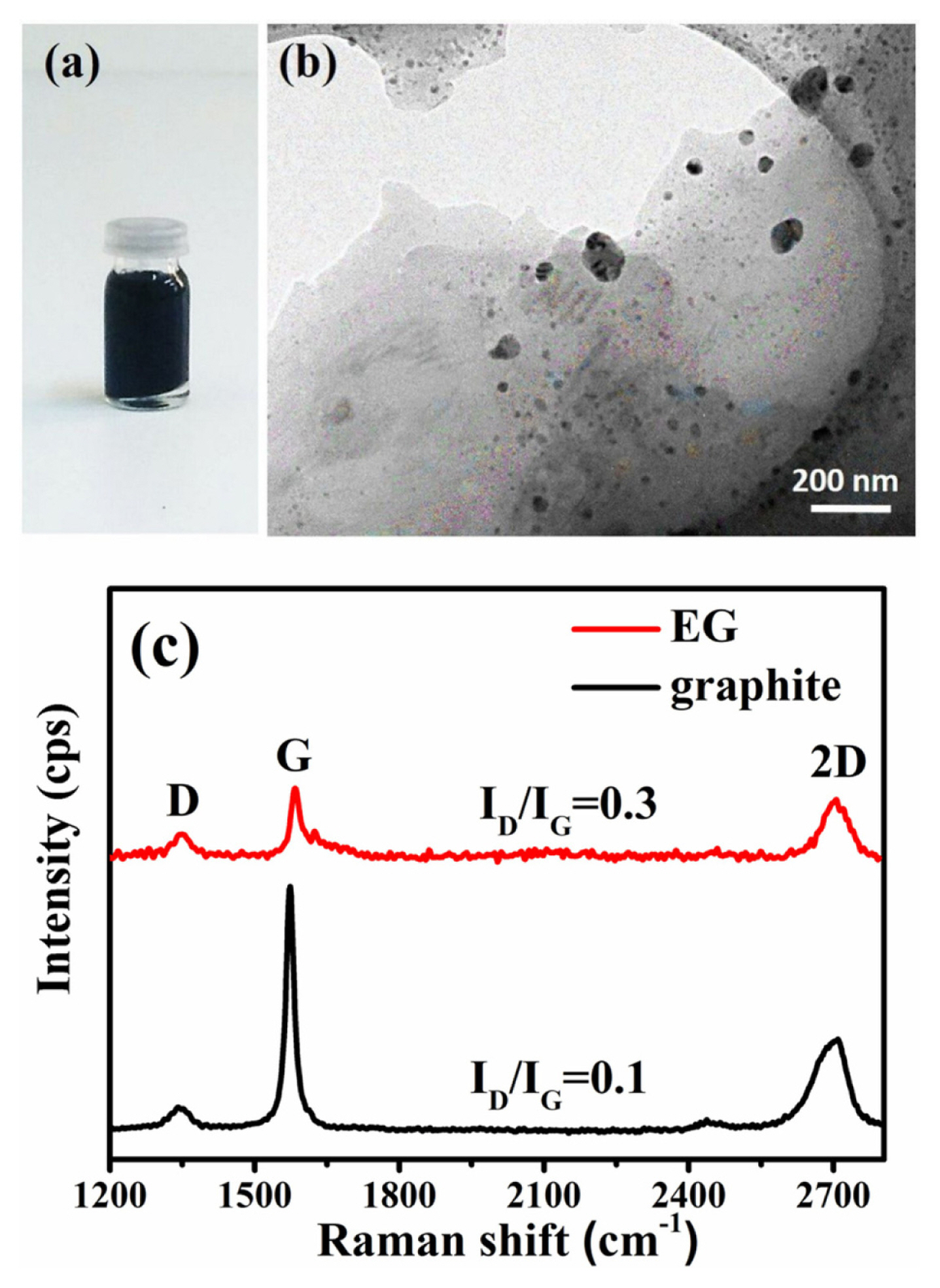
In liquid phase exfoliation, surfactant molecules lower the surface tension of water to overcome Van der Waals forces between graphene sheets. Moreover, CTAB molecules adsorbed onto the electron-rich graphene sheets create electrostatic repulsions and prevent re-stacking [18]. Fig. 1a demonstrates the digital image of the highly stable surfactant-stabilized graphene dispersion taken one year after its preparation. The TEM image of the dispersion reveals overlapping few-layer graphene (FLG) sheets (Fig. 1b). Complementary in situ Raman spectroscopy studies were performed to verify the formation of FLG (Fig. 1c). Low-intensity D band (at ~1350 cmŌłÆ1) observed in both graphite and graphene spectra can be ascribed to A1g breathing mode of hexagonal rings and result from the basal plane or edge defects. The G band emerges from first-order scattering of E2g phonons that involves in-plane bond-stretchings of sp2 carbons. The intensity of the G band decreases and shifts to a higher frequency (from 1573 to 1585 cmŌłÆ1) going from graphite to exfoliated graphene (EG), as less sp2 carbons are detected with decreasing number of graphene layers [19]. The number of graphene layers are reported to be less than 5 when the intensity ratio of 2D to G band (I2D/IG) is equal to or greater than one [20]. FWHM (full-width at half-maximum) of the 2D band is reported to be less than 30 cmŌłÆ1 for single-layer graphene and increase to 65ŌĆō70 cmŌłÆ1 for FLG [19]. EG showed 2D band characteristics typical of few-layer graphene with a FWHM of 70 cmŌłÆ1 (from Lorentzian fit) and a I2D/IG value of 0.8 [19,21]. Liquid phase exfoliation method utilized here allowed production of graphene sheets with a lower number of defects (defect ratio=ID/IG=0.3) compared to chemically reduced graphene oxides (rGO) reported in the literature (ID/IG= 1.2 ŌĆō 1.5) [22,23].
Exfoliated graphene (EG) sheets were used as a sacrificial template to induce the growth of birnessite-type MnO2 nanoparticles under hydrothermal conditions. The C atoms of EG sheets reduce KMnO4 (Mn+7) to Mn+4 and initiate the nucleation of MnO2 on the graphene surface: 4KMnO4 + 3C + H2O ŌåÆ 4MnO2 + K2CO3 + 2KHCO3. The KMnO4-limited hydrothermal reaction proceeds faster and dominates the growth of the MnO2 nanosheets: 4KMnO4 + 2H2O ŌåÆ 4MnO2 + 4KOH+ 3O2 [8,24]. The graphene-templated growth of MnO2 nanosheets proceeds along the ab plane and leads to formation of layered birnessite-type MnO2 [8]. As revealed by SEM images (Fig. 2a, b), MnO2 nanosheets with a thickness of 10 nm assemble into flower-like nanospheres (200ŌĆō300 nm in diameter) forming a hierarchical morphology which promotes efficient electrode-electrolyte interactions and fast transport of ions. TEM images (Fig. 2c, d) reveal lattice spacing of 0.24 nm corresponding to (101) plane of birnessite-type MnO2. Lattice fringes in different orientations indicate the polycrystalline nature of MnO2 in line with the SAED pattern.
The XPS survey spectrum (Fig. 3a) confirms the existence of Mn and O elements in the MnO2 sample. The trace K 2p and C 1s peaks in the spectrum result from stabilizing interlayer K+ cations and residual graphene template, respectively. The spin-induced energy separation between Mn 2p1/2 and Mn 2p3/2 peaks was calculated as 11.7 eV, confirming the oxidation state of +4 for Mn (Fig. 3a- inset) [25,26]. The three oxygen peaks detected in the O 1s spectrum (Fig. 3b) can be assigned to the lattice oxygens in [MnO6] unit cells (527.4 eV), the Mn-OH (528.9 eV), and H-O-H groups (529.9 eV) [27].
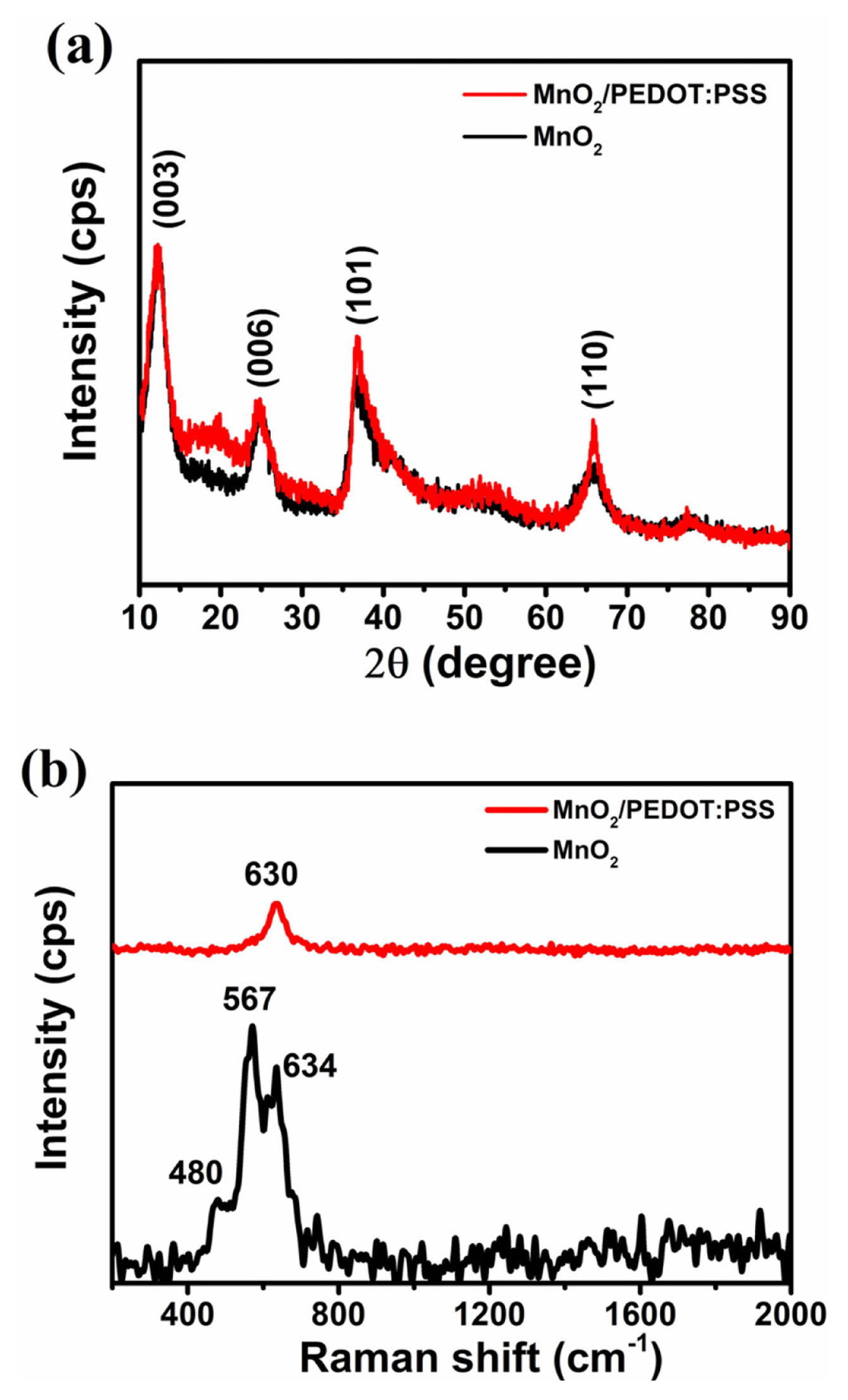
Graphene-templated MnO2 nanostructures were coated with a thin conductive PEDOT:PSS layer to enhance capacitive performance. The four XRD peaks at 12.3┬░, 24.8┬░, 36.6┬░ and 65.5┬░ observed both for the MnO2 and MnO2/PEDOT:PSS samples (Fig. 4a) can be indexed to hexagonal birnessite-type MnO2 [28]. The 7.170 ├ģ interlayer space (d003) indicates the birnessite structure consisting of stacked monolayers of edge-shared [MnO6] octahedra intercalated by K+ cations and H2O molecules. The additional broad peak at 19┬░ with a d-spacing of 4.62 ├ģ observed in MnO2/PEDOT:PSS pattern is attributed to ŽĆ-stacked PEDOT:PSS [29]. In the Raman spectrum of MnO2 (Fig. 4b), three characteristic MnŌĆōO lattice vibrations of birnessite-type MnO2 were observed at 480, 567, and 634 cmŌłÆ1 [30]. In the MnO2/PEDOT:PSS sample, MnŌĆōO lattice vibrations merged into one broad peak at 630 cmŌłÆ1 as a result of thin PEDOT:PSS coating.
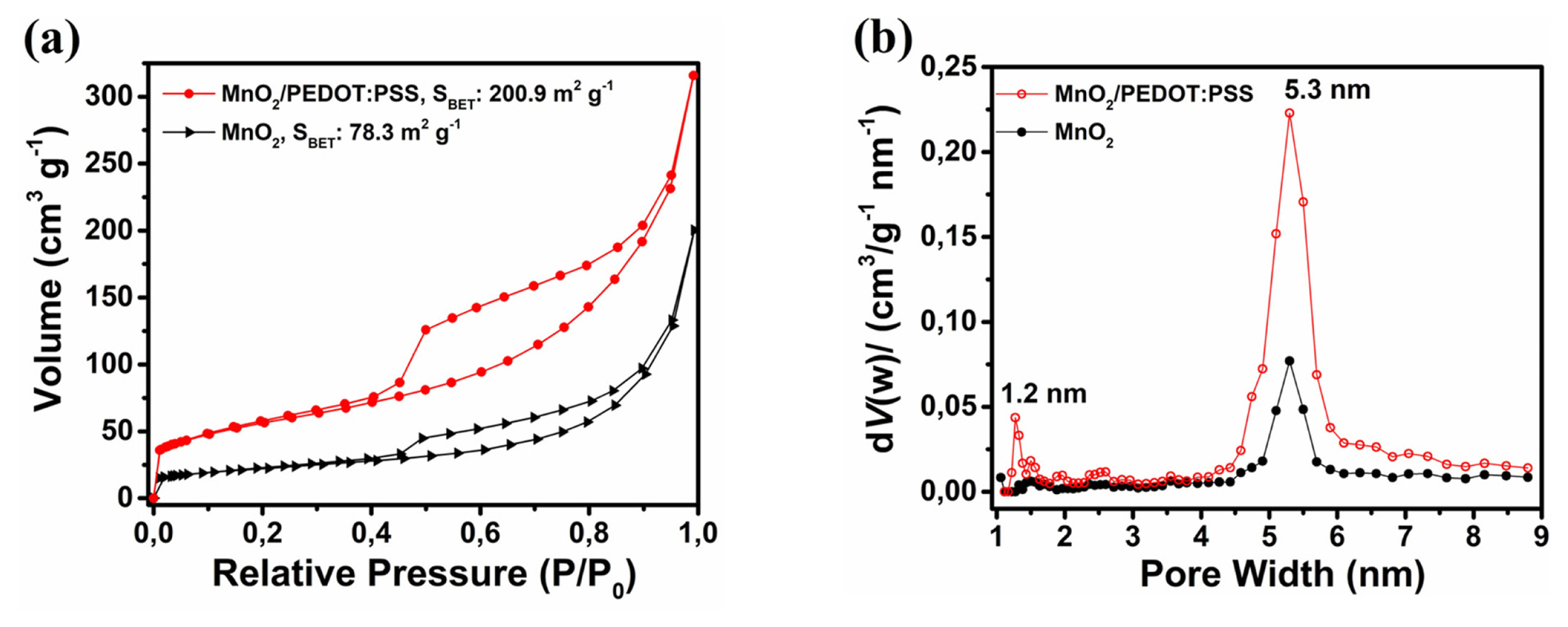
Specific surface area and porosity of the samples were investigated by Nitrogen (77 K) gas-sorption measurements (Fig. 5). According to IUPAC classification [31], both samples show Type IV isotherm with an H3 type hysteresis indicating mesoporous structure (Fig. 5a). The higher amount of adsorption at low relative pressures (P/P┬░<0.1) observed for MnO2/PEDOT:PSS indicates the presence of micropores and the larger width of the hysteresis indicates higher volume of mesopores. Both samples display narrow pore size distribution with peak maximums located at 5.3 nm while MnO2/PEDOT:PSS has an additional low-intensity peak at 1.2 nm (Fig. 5b). PEDOT:PSS coating increases the BET specific surface area from 78.3 m2gŌłÆ1 to 200.9 m2gŌłÆ1 and the corresponding pore volume from 0.09 cm3gŌłÆ1 to 0.24 cm3gŌłÆ1. Hierarchically porous MnO2/PEDOT:PSS promises higher capacitance as micropores (< 2 nm) provide additional surface for electrode-electrolyte interactions and high rate capability as mesopores (2ŌĆō50 nm) provide fast ion transport during charge-discharge.
3.1 Electrochemical studies
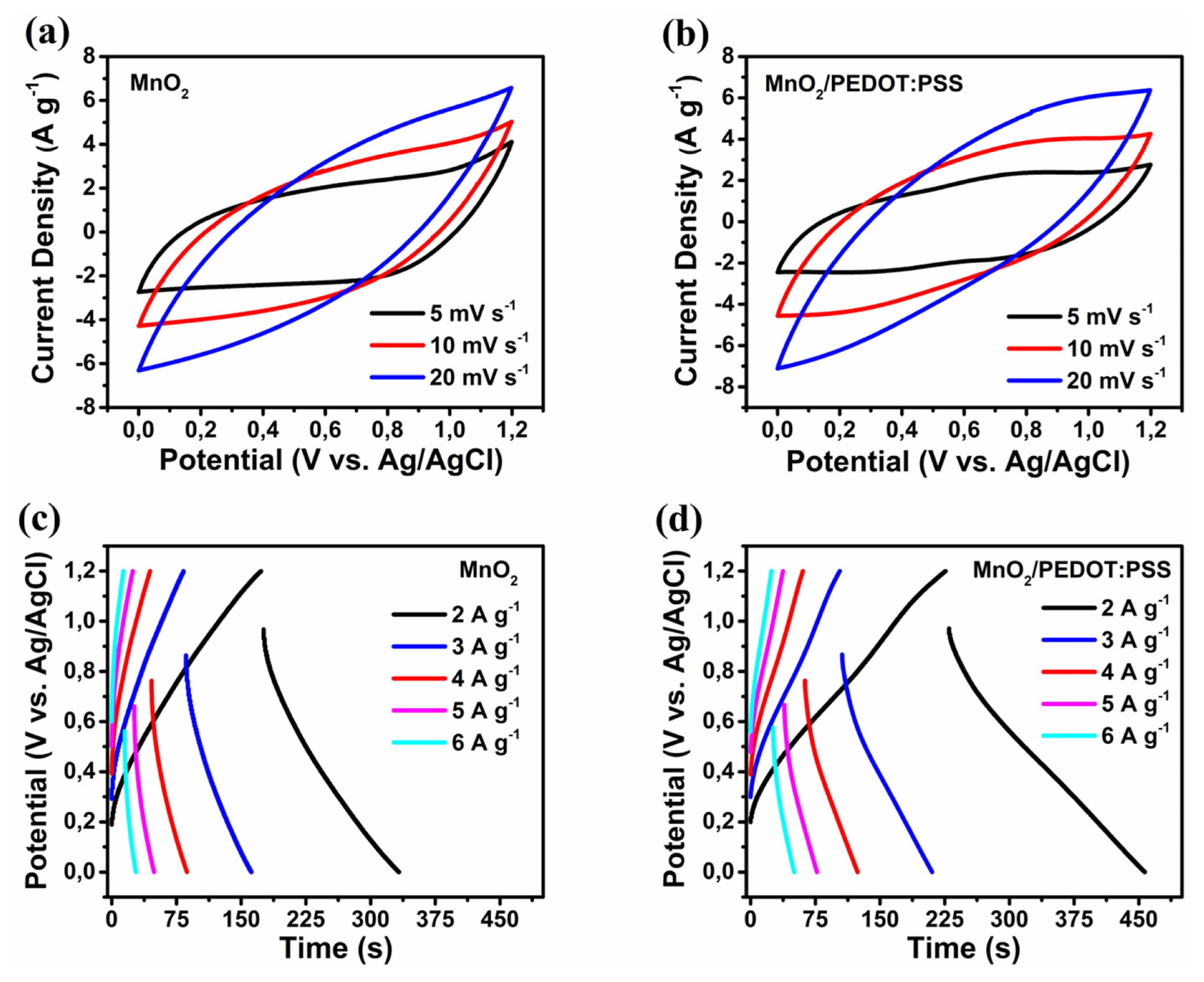
Energy storage capabilities of the MnO2 and MnO2/PEDOT:PSS samples were investigated by cyclic voltammetry (CV) and galvanostatic charge-discharge (GCD) studies in 1M Na2SO4 aqueous electrolyte (0 ŌĆō 1.2 V vs. Ag/AgCl). Slightly larger CV areas (Fig. 6a, b) and longer discharge times (Fig. 6c, d) observed for MnO2/PEDOT:PSS indicate higher capacitance compared to MnO2. Part of this capacitance results from the surface redox reactions between MnO2 and alkali metal cations (Na+ in this case) or protons (H+): MnO2 + xNa+ + yH+ + (x + y) eŌłÆ Ōåö MnOONaxHy [32]. Highly conjugated PEDOT:PSS with high electron mobility improves surface conductivity while PEDOT redox couple aids in overall pseudocapacitance: PEDOT+. PSSŌłÆ + xNa+ + yH+ + (x+y) eŌłÆ Ōåö PEDOT0. PSSŌłÆ. NaxHy [33]. The deviation of voltammograms from the standard rectangular shape and the initial voltage (IR) drops at discharge result from diffusion limitations of surface redox reactions.
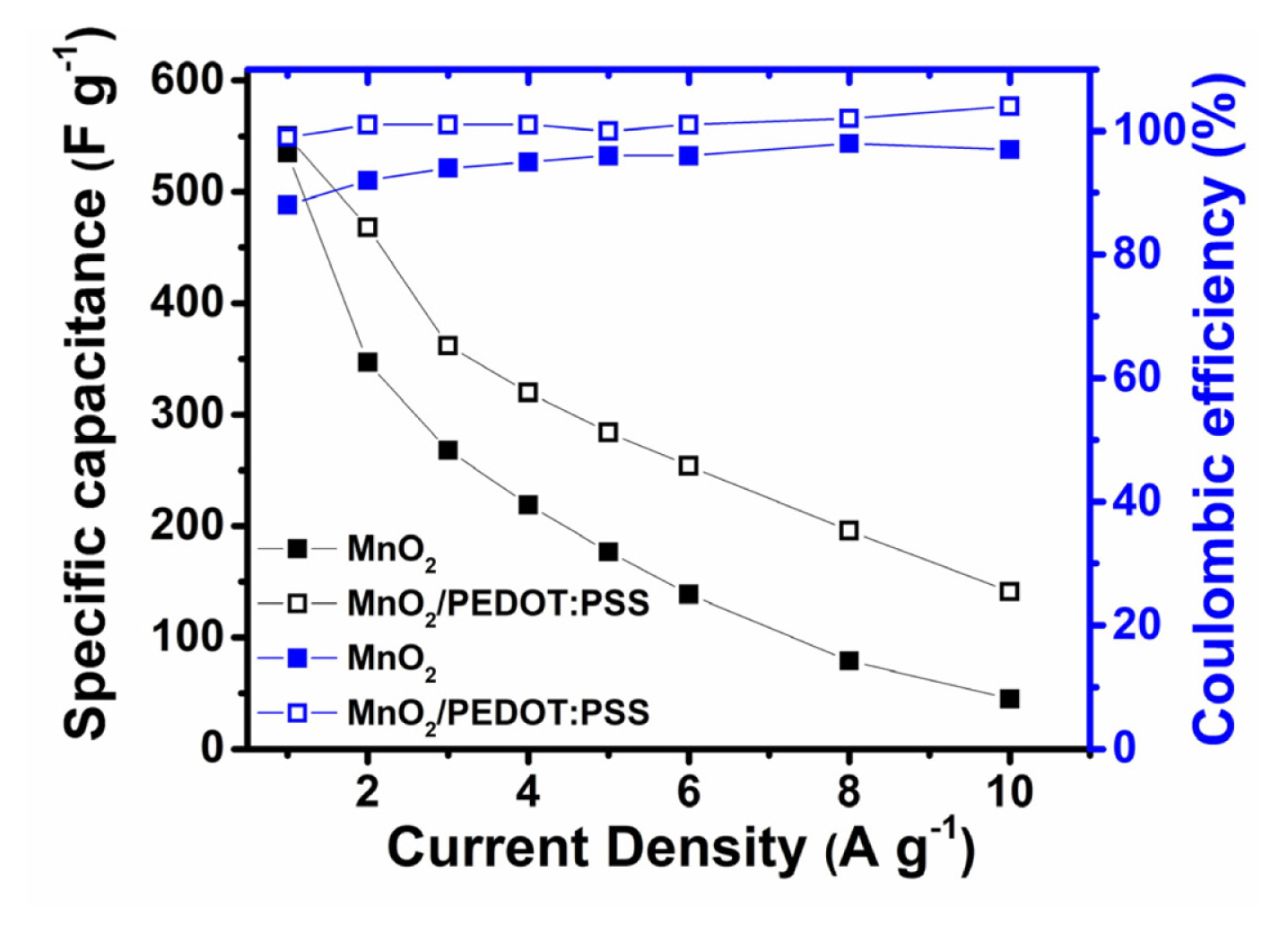
The specific capacitances of the samples are calculated at different current rates (1 to 10 A gŌłÆ1) and plotted in Fig. 7. Both MnO2 and MnO2/PEDOT:PSS provide high specific capacitances at 1 A gŌłÆ1 (535 and 550 F gŌłÆ1, respectively). PEDOT:PSS polymer coating increases the conductivity and thus coulombic efficiency of MnO2 from 88% to 99% at 1 A gŌłÆ1. Higher rate capability of MnO2/PEDOT:PSS compared to MnO2 (141 vs. 45 F gŌłÆ1 at 10 A gŌłÆ1) results from higher volume of mesopores which enable fast and easy transport of charge carriers. MnO2 and MnO2/PEDOT:PSS samples prepared in this work delivered superior capacitances and rate capabilities compared to previously reported graphene templated MnO2, MnO2/rGO and MnO2/rGO/conductive polymer nanocomposites prepared by harsh chemicals and laborious methods [8ŌĆō11,13,16,34].
3.2 Asymmetric Supercapacitors
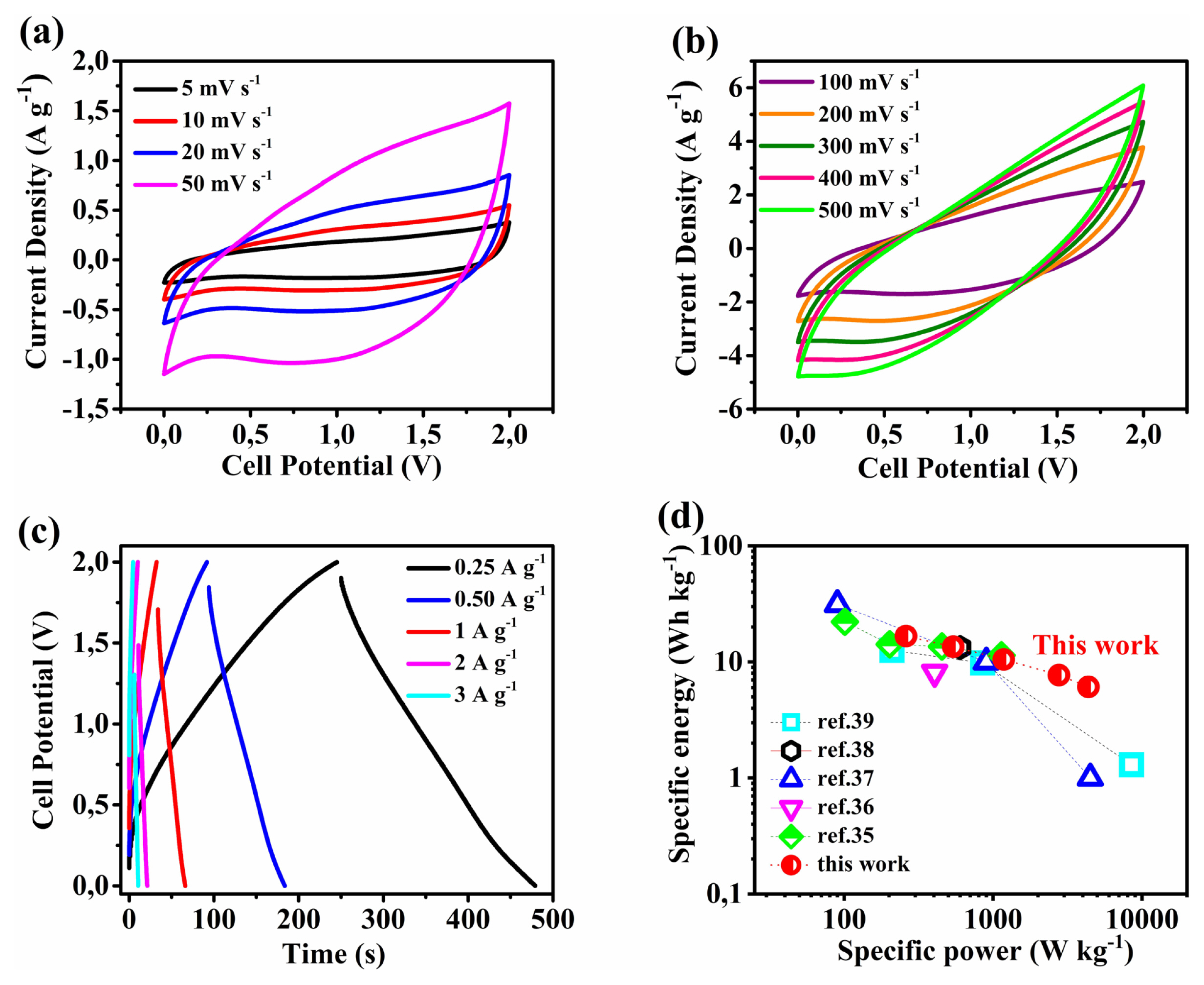
Asymmetric supercapacitors (ASCs) were assembled with Fe3O4/Carbon nanocomposite negative electrode against MnO2 or MnO2/PEDOT:PSS positive electrodes. The CV and GCD curves of the devices are recorded in a 1M Na2SO4 aqueous electrolyte (╬öV= 2 V). The Fe3O4/C//MnO2/PEDOT:PSS device delivered higher capacitive performance at 1A gŌłÆ1 charge-discharge rate (Cs = 21 F gŌłÆ1, E = 12Wh kgŌłÆ1, and P = 1.2 kW kgŌłÆ1) compared to the Fe3O4/C//MnO2 device (Cs = 16 F gŌłÆ1, E = 9 Wh kgŌłÆ1, and P = 1.1 kW kgŌłÆ1). As shown in Fig. 8 the Fe3O4/C//MnO2/PEDOT:PSS device exhibits a maximum specific energy of 18 Wh kgŌłÆ1 (0.25 A gŌłÆ1) and a maximum specific power of 4.5 kW kgŌłÆ1 (3 A gŌłÆ1). The Ragone plot (Fig. 8d) compares the performance of MnO2/PEDOT:PSS device obtained in this work to previously reported MnO2-based asymmetric supercapacitors and proves better rate capability of the device [35ŌĆō39].
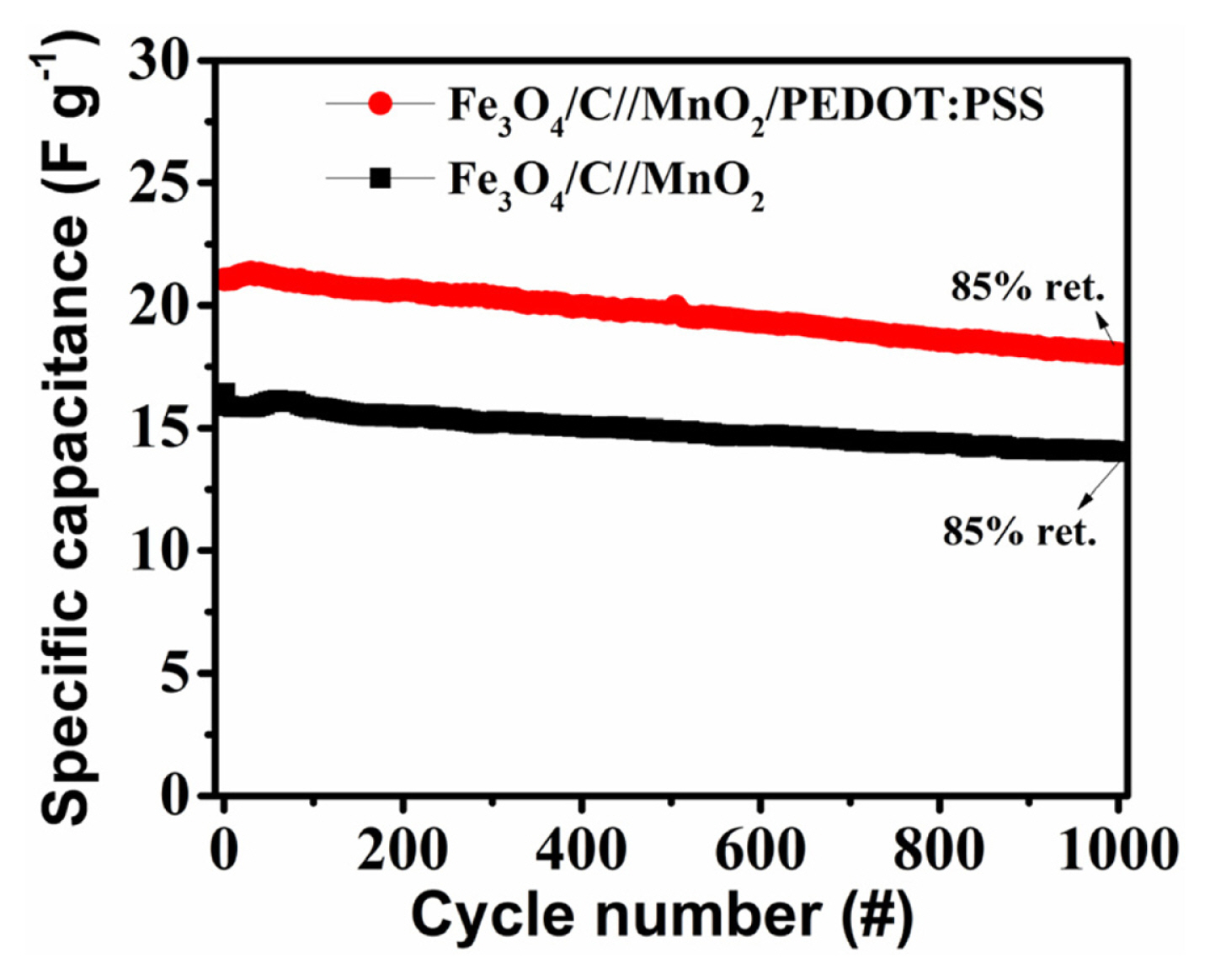
The Fe3O4/C//MnO2 and Fe3O4/C//MnO2/PEDOT:PSS asymmetric supercapacitors both remain chemically and structurally intact over the span of 1000 charge-discharge cycles at 1 A gŌłÆ1 and retain 85% of their initial capacitances (Fig. 9). SEM-EDS mappings of MnO2 and MnO2/PEDOT:PSS electrodes reveal only 5% active material loss (Mn) after 1000 cycles (Fig. 10) and prove that the open network of MnO2 enabled by graphene templating prevents microstructural degradation upon continuous charge-discharge.
4. Conclusions
In summary, a simple approach has been demonstrated for producing a high performance nanocomposite electrode consisting of exfoliated graphene-induced ╬┤-MnO2 and PEDOT:PSS (MnO2/PEDOT:PSS). A few-layer graphene was produced by a safe and scalable liquid-phase exfoliation (LPE) method and used as a sacrificial template for hydrothermal synthesis of birnessite-type hierarchical manganese oxide (╬┤-MnO2). Graphene templating delivered a chemically and structurally intact MnO2 with a high capacitance and electrochemical cycling stability. Conductive PEDOT:PSS coating further improved the capacitance and rate capability. High performance MnO2/PEDOT:PSS composite electrode obtained by scalable and environmentally friendly methods promises for future energy storage applications.