1. Introduction
Recent high demands for new high energy-efficient systems such as electric vehicle and energy storage systems require cost-effective rechargeable batteries because they are huge, unlike the conventional applications of mobile phones and lap-top computers [1–3]. Since 1991, when lithium ion batteries (LIBs) were firstly commercialized, high electrochemical performance of the active materials to reversibly accommodate lithium ions during electrochemical cycling has been vigorously investigated [4–8]. Among those active material candidates, LiMn2O4 having spinel structure has several apparent merits; i) environmentally friendliness ii) cost-effective, iii) fast lithium diffusion through 3 dimentional lithium ion channels and iv) full lithium ion utilizations without degradation of crystal structure [9–12]. However, the unfavorable Mn dissolution from LiMn2O4 via Jahn-Teller distortion and the surface electrolyte decomposition are still crucial problems to limit their practical applications by reducing cycle life and increasing polarizations [13–17]. To solve these issues, the several doping methods and the various surface coatings have been investigated by many publications [18–21]. The surface-modification is a very ready-to-use approach with the commercialized battery active materials which was carefully synthesized with or without doped elements [22–25]. For practical applications, several engineering factors should be considered to deliver a proper morphology of powder. A lot of conventional surface coating materials for the active materials are focused on the electron insulator and the chemically stabilizer. To reduce the electrochemical degradation of the electrolyte on the cathode materials, it is a very good approach to block electron transportation by using an electron-insulating coating. Unfortunately, this passivation film limits power density because the passivation for electron barrier hinders lithium ion transportation as well as electron transportation. Despite the electron-insulating surface coating is beneficial to reduce the electrolyte decomposition, the unavoidable high ionic transfer resistance through the layer is not beneficial for high rate capability. In this regard, it is controversial to conclude that the electronic insulating inorganic material coating should be pursued for LIBs. Therefore, highly ionic conducting materials such as Li7-La3Zr2O12(LLZ) and Li1.3Al0.3Ti1.7(PO4)3(LATP) are investigating by many researchers [26–29]. However, the highly lithium ion conducting materials are very restricted for the surface coating on the active material because of their complexity of the synthesis, requiring high temperature and huge pressure which harms the crystal structure of the active materials. In this report, a new ionic conductive surface coating by using a room temperature synthetic process for Li2TiF6 having a lot of lithium channels inside is suggested as a coating material for LiMn2O4. Li2TiF6 is well known as a lithium conductive material. [30,31] We evaluated the battery performance with Li2TiF6 coated LiMn2O4 cathode material in respect of cycle performance and rate capability in comparison with the pristine LiMn2O4 to investigate the effect of the coating.
2. Experimental
2.1. Materials Synthesis and Characterization
Li2TiF6 was coated on the LiMn2O4 (Aldrich) with a facile way of co-precipitation by using the mixture of Li2CO3 (Aldrich) powder and H2TiF6 solution (Aldrich). Firstly, LiMn2O4 powder was mixed with distilled water and Li2CO3. Then the stoichiometric amount for Li2TiF6 of 60 wt.% aqueous solution of H2TiF6 was slowly added into the slurry having LiMn2O4 and Li2CO3 and completely mixed by pestle and mortar. And the obtained mixture was thoroughly dried at 60°C for 24 hours in convection oven. For these processes, the amounts of the coating materials were controlled to 1, 2, 5 and 10 wt% of LiMn2O4. Subsequently, Li2TiF6 coated LiMn2O4 powder was analyzed by an inductively coupled plasma-mass spectrometer (ICP-MS, ELAN 6100) to confirm the ratio of the Li2TiF6 with LiMn2O4 as well as the presence of Li2TiF6. Those prepared active materials were identified with X-ray diffraction (XRD, Rigaku, D-MAX2500-PC, Japan) using Cu Kα radiation (λ = 1.5412 Å, 40 kV, 100 mA). And the morphologies of prepared materials were detected by using transmission electron microscopy (TEM, LIBRA 120, Germany) and Field-Emission Scanning Electron Microscope (FE-SEM, SUPRA 55VP, Germany).
2.2. Electrochemical Measurements
The electrode was composed of the cathode active materials (80 wt. %), super P (conducting agent, 10 wt. %, Timcal), and polyvinylidene fluoride (PVDF, binder, 10 wt. %, Solef 6020). Those materials were homogeneously mixed with N-methyl-2-pyrrolidinone (NMP, Aldrich), and the slurry was bladed on an aluminum foil. The obtained electrode sheets were dried to remove the NMP solvent and the residue moisture at 60°C under vacuum condition for 8 hours. Before using the electrode, the electrode sheet was roll-pressed. The electrode sheets were punched to round electrodes having a diameter of 1.1 cm.
The 2032 type coin cells were assembled as half cells and lithium foil was used as the counter electrode. The electrolyte was composed of 1 M LiPF6 and ethylene carbonate/diethyl carbonate (EC:DEC = 1:1, v/v). And all the processes for coin-cell fabrication was carried out in an argon-filled glove box to eliminate possible moisture impurities. Galvanostatic electrochemical experiments were performed by using a WBCS3000 cycler (Won-A tech, Korea). Charging and discharging procedure of the cells was performed in the potential range between 3.0 and 4.3 V at various current conditions. Also, the electrochemical impedance spectroscopy (EIS) data were analyzed by using the 2032 coin cells and the electrochemical workstation (Autolab) at room temperature (25°C). The amplitude was 5 mV and the frequency range was from 10 kHz to 10 mHz. Before measurement of EIS, the cell was rested for an hour to stabilize the potential of the cell.
3. Results and Discussion
3.1. Preparation and Characterization of Li2TiF6-LiMn2O4
Before the coating of LiMn2O4 with Li2TiF6, the morphology of pristine LiMn2O4 (Aldrich) without additional treatments was observed by SEM (Fig. 1a and b). Several researches have been reported on the synthesis of porous LiMn2O4. [32,33] The particles have secondary particles having μm scale diameters. The secondary particles are comprising with primary particles having tens nm scale diameter. The structure of primary particles is almost octahedral with diameters of 10~30 nm. These primary particles are packed into 300~400 nm secondary particles. These LiMn2O4 particles were modified by coating process. The hierarchical structure has induced internal meso and macro size pores. Fig. 1c and d shows the information of pores constructed in the hierarchical LiMn2O4. The N2 adsorption curve in figure 1c demonstrated that small amount of the meso pore was formed and the major pore was composed of the macro-size pore. In Fig. 1d, from calculation, we confirmed that the BET surface area is 150 m2/g−1 and the pore size is in the range of 10~80 nm. Especially, the major size of meso pore is of 10 to 50 nm.
The coating process of Li2TiF6 is very simple and facile. Li2TiF6 is synthesized with H2TiF6 and Li2CO3 according to the following reaction.
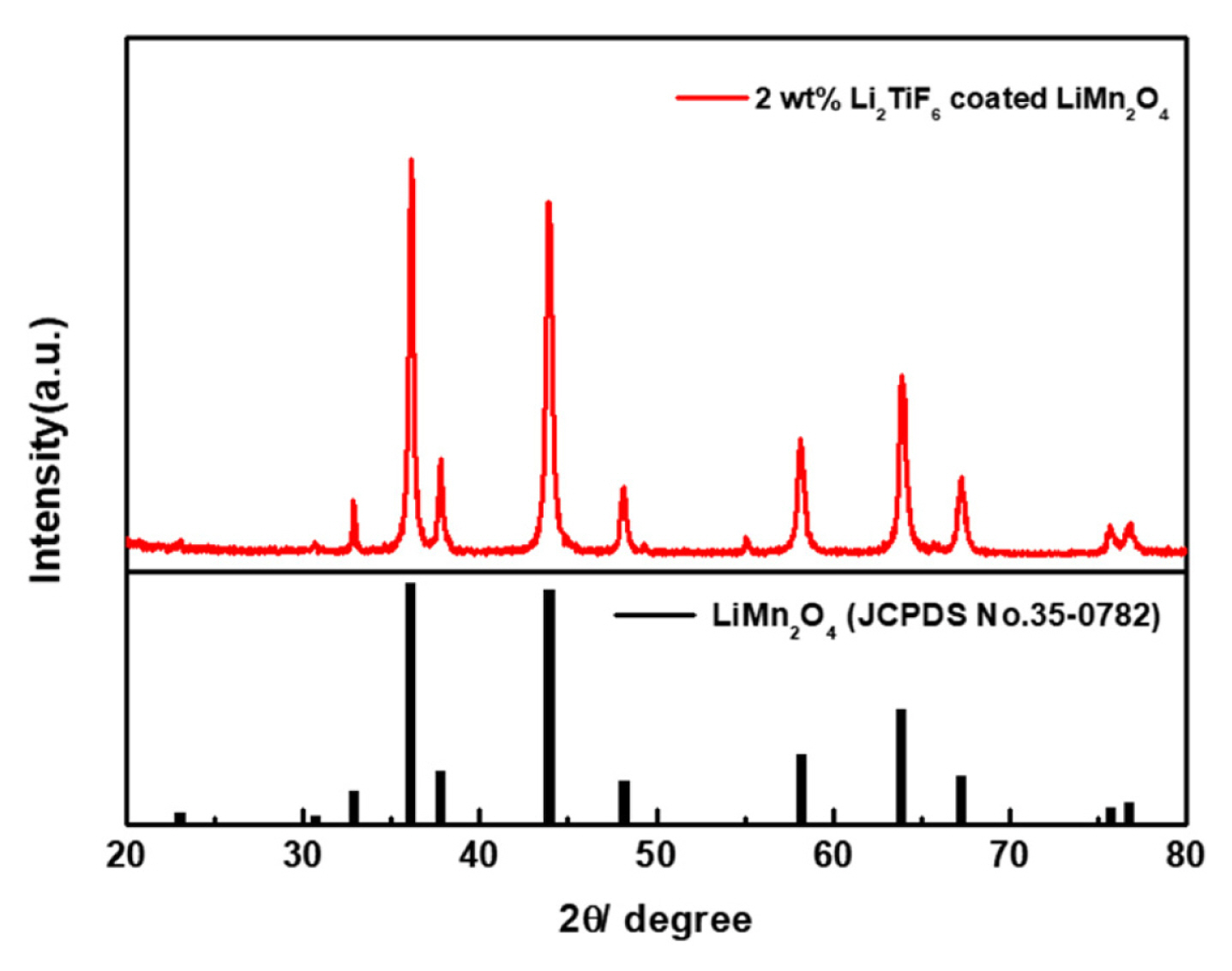
The reaction finished within just few seconds generating the CO2 and water. During the co-precipitation of Li2TiF6, the gas evolution of CO2 was observed as small bubbles. To certify the coating effects on the structural behavior of LiMn2O4, XRD analyses were performed with the surface coated LiMn2O4. As the Fig. 2 shows that the crystalline structures of the LiMn2O4 had not been significantly changed after the coating process. So, it can be confirmed that the active materials of LiMn2O4 was highly preserved after the coating process.
On the other hand, the coating material of Li2TiF6 was not observed by XRD pattern, because the coating amount is not enough to make a highly ordered morphology for being observed by XRD. Consequently, the ratio between Li2TiF6 and LiMn2O4 was analyzed by using ICP. Analyzing the portion of ions for the prepared powder in the Table 1. It showed that the amount of the coating materials of Li2TiF6 were calculated on the mass of LiMn2O4 as we designed. As the total amount of Li2TiF6 coating increases, the portion of Ti also increases.
The morphologies of Li2TiF6-LiMn2O4 composite were confirmed by TEM in Fig. 3. In Fig. 3a, the TEM image of the raw LiMn2O4 does not have the additional layer on the surface. In Fig. b, c and d, the surface of LiMn2O4 has a bright film which is indicated by black arrows and distinguished from the LiMn2O4. It is thought to be Li2TiF6 coating on the surface. The TEM images show that the coating thickness was very thin and varied from 5 to 20 nm at each ratio. It is found that the surface coating of Li2-TiF6 is very homogeneous on the surface as shown in EDX image in Fig. 3 because the atomic distribution of Ti is very wide and homogeneous. After coating 10 wt%, the average thickness was about 20nm and the coating formation was uneven and rough. It can be considered that more than 10wt% coating can have negative effect on the electrochemical performances due to the uneven and thick coating layer.
3.2. Electrochemical Performance
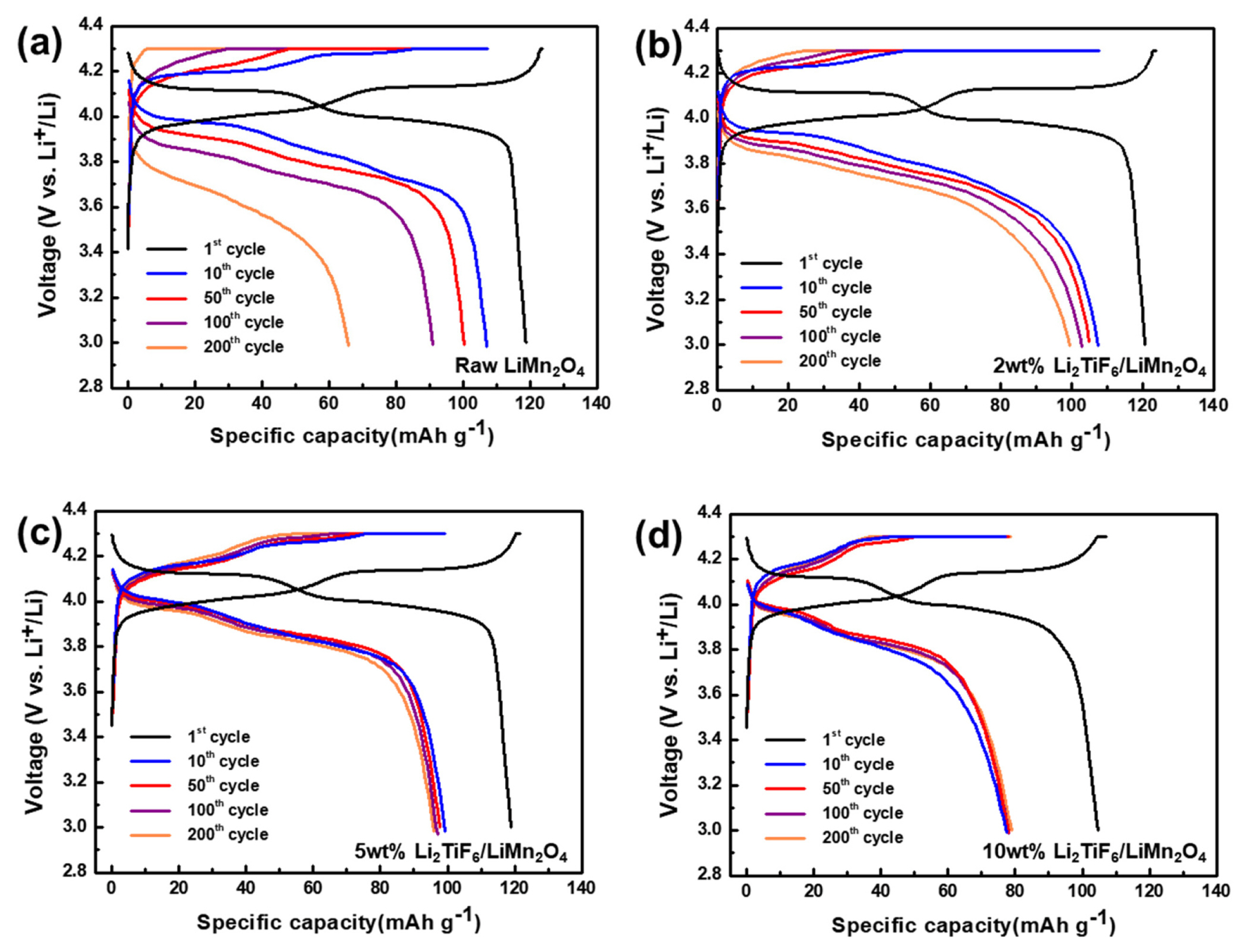
The electrochemical performances were evaluated to discuss for suppressing the side reactions by the surface protection layer. The cycleability of the cells having different types of positive electrodes with 0, 2, 5 and 10 wt.% of Li2TiF6 at room temperature over 200 cycle is describe in the Fig. 4a. All the cells had passed the formation cycle (initial cycle, 0.1c) which can stabilize the cathode materials at high voltage range. The cell with the raw LiMn2O4 exhibited a distinct capacity decaying behavior during the overall cycles. Initially it has a specific capacity of 106.9 mAh g−1 but 91.0 and 65.7 mAh g−1 for 100th and 200th cycle. In the long term cycle performances, this unfavorable surface film as the by-product stands out as a negative effect. But the specific capacities of all the Li2TiF6-LiMn2O4, regardless of the coating thickness, were highly preserved. For instance, the specific capacity of the 2wt% Li2TiF6-LiMn2O4 is 102.9 mAh g−1 after 100 cycles and 99.4 mAh g−1 after 200cycles. The capacity retention is just 61.4% for non-coated LiMn2O4 after the 200th cycle, but that is 96.6% at 2 wt% Li2TiF6-LiMn2O4. On the other hand, the cell with 10 wt% Li2TiF6-LiMn2O4 exhibited a low capacity of 75.0 mAh g−1 at the first cycle after the formation cycles. With the TEM images in figure 3d, it is considered that the coating layer is too thick to pass lithium ions well into the materials. Despite of the initial low capacity, the capacity retention is highly preserved in the cycle life from the coin cell with 10 wt% Li2TiF6-LiMn2O4. The surface coating of Li2TiF6 is still effective to stabilize the cycle life. Considered capacity values and capacity retention, the optimization ratio of the Li2-TiF6 is 2wt%. It suggests that the coating deters the generation of poor surface films from side reaction between electrolyte and surface of cathode materials [34].
The coating layers prohibited that the electrolyte directly contacts with electrode materials. It reduces the surface polarization via avoiding the electrolyte decomposition on the surface. These can be also considered the rate capability tests at difference rate (1,5, 10, 20, 30, 40, and 50 C) in figure 4b. It is found that the capacity reductions were showed when the c-rate increases. But the different electrodes have different reduction behaviors in Fig. 4b. Especially, the 2wt% Li2TiF6-LiMn2O4 displayed the highest rate capability among the cells with the prepared samples. This result is highly correspondent with the Fig. 4a, because of that the coating thickness is well optimized for the capacity and retention [35,36]. If the layer thickness is too thick, it will impede the lithium movement when the lithium ion passes through the boundary. The combination of the preventing from side reaction and the deterrent of the lithium ion movement leads to the optimization of Li2TiF6 ratio. So, when using the lithium conducting materials for the passive layers on the active materials’ surface, it leads to the lithium moving easier and finally make it possible to work in very high-rate charge and discharge states without deterioration of capacity. The coating layers on the cathode materials can be a protection that deters the side reaction between electrolyte and cathode materials. The advantages of coating process are widely known and lots of materials such as, Al2O3 and ZrO2, have already been applied to the coating materials [24,37]. But these materials are low lithium conductive materials and synthesis process consist of several heat-treatment processes. It is well known that the coating layer can prohibits the acidic electrolyte attacks on the active materials causing the by-product such as, carbonate, oxygen gas, and Mn dissolution [38,39]. For all that, the low lithium conductivity can disturb the kinetic of lithium ion passing through the coating layer especially high rate. Most of the coating materials are low lithium conductive materials. Those candidates have a strong effect on the spinel materials which support the high rate performance due to the 3 dimensional lithium ion conducting channels [40]. For this reason, lithium conductivity of the coating materials is important for the spinel materials coating.
The voltage profiles were obtained from the cycle performances for the 1st, 10th, 50th, 100th, and 200th cycles. Based on the equilibrium potential of 4.1 V vs. Li/Li+ for electrochemical reaction for lithium ion accommodations in LiMn2O4, the charge and discharge charge curves have polarizations which are originated from the various resistance in the cells. In Fig. 5a for the cell of the raw LiMn2O4, the polarization continued to increase along the cycle number. For the raw LiMn2O4, the resistance was affected by by-products which were formed by Mn dissolution and electrolyte decompositions during cycling [41]. The first reduction peak shifted from 3.98 V at 2 cycles to 3.91 V at 50 cycles and 3.84 V at 100 cycles. On the other hand, as shown in Fig. 5b, c and d, the coating reduces this unfavorable high polarization during cycling. On the other hand, the peaks of Li2TiF6-LiMn2O4 cells are rarely shifted regardless of the cycles. For the 2 wt% Li2TiF6-LiMn2O4, the first reduction peaks are 4.02 V at 2 cycles, 3.98 V at 50 cycles and 3.96 V at 100 cycles. In addition, 5 and 10wt% Li2TiF6-LiMn2O4 in Fig. 5c and d, the first reduction peaks are 3.98V at the 1st cycle. After the 50th cycle, the peaks are rarely shifted from 3.95 V by 200 cycles. So, it is confirmed that the resistance depending on the formation of by-product which comes from side reaction between electrolyte and raw LiMn2O4 is effectively compressed.
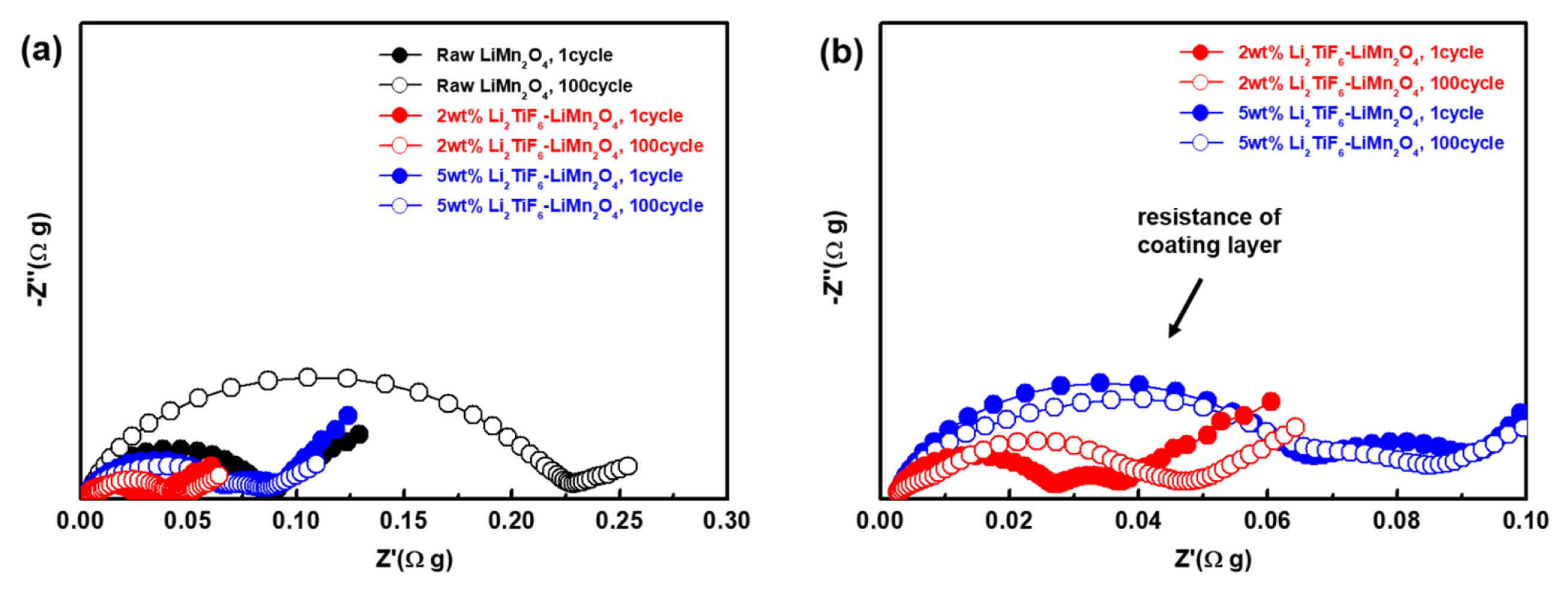
To investigate the resistance for the cells, the electrochemical impedance spectra (EIS) tests were performed. Fig. 6 shows the EIS of the cells with each of the coating 0, 2 and 5 wt% after 1, 50, 100 cycles. To the compensation for the weight, the impedances were multiplied with electrode weight. Every Nyquist plot has a semi-circle of which diameter is thought to be surface resistance. The semi-circle of raw LiMn2O4 after 1 cycle is larger than others in high frequency. And the radius of semi-circle is increased to 0.225 Ωg (Z′). Whereas, the diameter of the semi-circles of the Li2TiF6 coated cells are almost same even after the 100th cycle. These data indicate that the surface was highly preserved without severe deterioration between electrolyte and cathode materials, from side-reaction. Therefore, the Li2TiF6 layers effectively prevent the formation of resistance. And the semi-circle diameter of 2wt% Li2TiF6-LiMn2O4 is the lowest, which is fitted well the cell performances as noted above. It is because that the lithium conductive characteristics of Li2TiF6 mitigates the impact which comes from inserting lithium ion through the interface of LiMn2O4.
4. Conclusions
Highly ionic conducting Li2TiF6 coatings by a simple and facile method greatly improve the cell performance for long-term cycle life as well as rate capability. Regardless of using artificial passive layer, the capacity fading is not suppressed at high rate of charging and discharging operations. Because the non-lithium conductive materials impede the lithium ion transportation when the lithium ion pass through the interface from electrolyte to the active materials. Electrochemical performance was improved by using porous without coating of Li2TiF6. However, the electrochemical performance of non-coated LiMn2O4 deteriorated more that Li2TiF6 coated LiMn2O4 at high charging-discharging rate of 20C. Li2TiF6, lithium conducting materials, make it possible to bringing the long term stability even very high rate charging and discharging operation. From suppressing the formation of by-product from side-reaction, the stability is maintained after the 200th cycle. And the cycle life is greatly stabilized due to the lithium conducting materials. This study suggests that the Li2TiF6 coating contributes to the industry from the high-power cathode materials with a simple coating process.