


1. Introduction
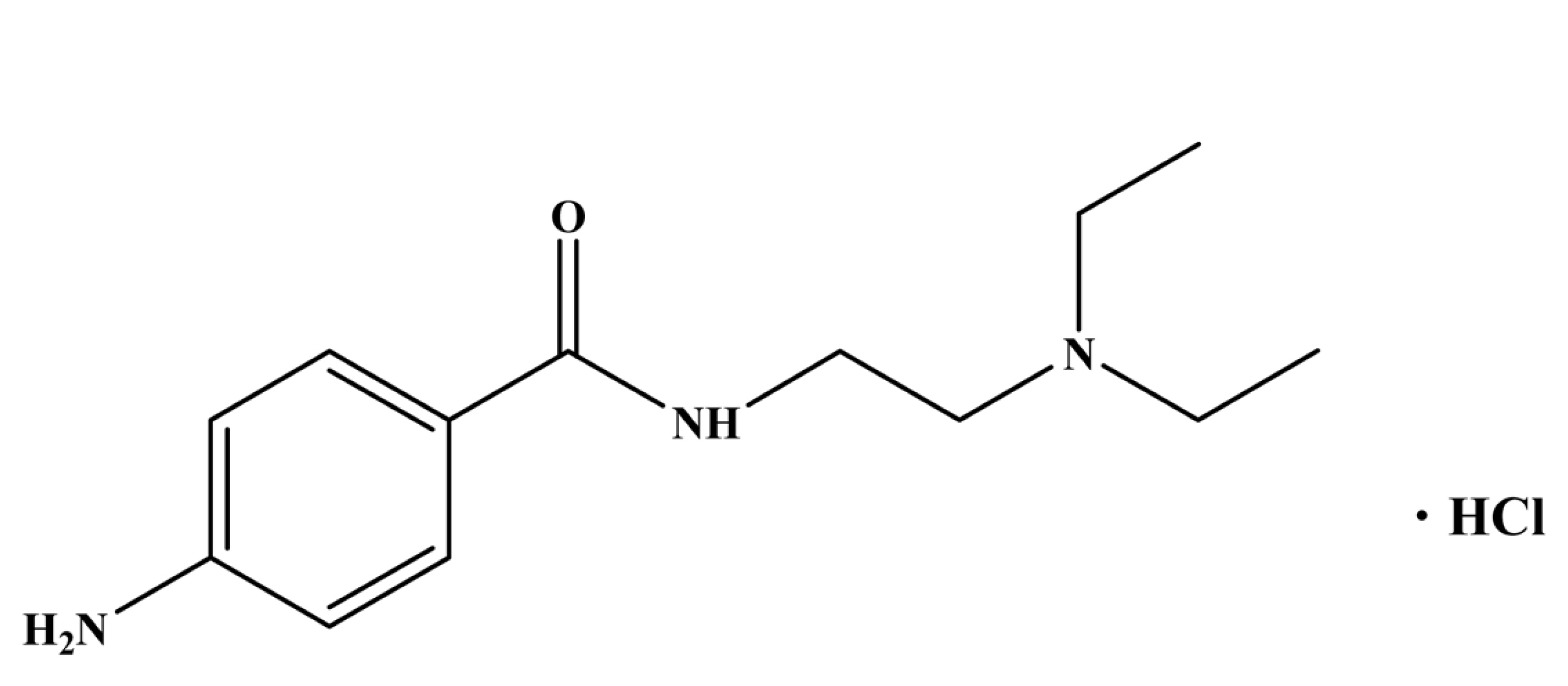
Antiarrhythmic drug therapy target towards restoring normal rhythm and conduction. Thereby, when it becomes impossible to revert back to the rhythm of normal sinus, drugs may be used to prevent more serious and possibly lethal arrhythmias from occurring [1]. Antiarrhythmic agents are prescribed to treat both auricular and ventricular arrhythmia and prevent their recurrence. It is observed that, procainamide and its main active metabolite, N-acetyl-procainamide (acecainide) with other antiarrhythmic drugs are found to be susceptible which can be clinically monitored in practices, especially in those patients who have slow kinetics of elimination due to their narrow therapeutic range and also tachycardia effects in an overdose [2]. Procainamide hydrochloride (4-amino-N-[2-(diethylamino)ethyl]benzamide;hydrochloride) as shown in Scheme 1 is the hydrochloride salt form of procainamide, an amide derivative which is known to exhibit class 1A anti-arrhythmic property. It is found to perform as a Na+ channel blocker of the cardiomyocytes [3]. Citation [4] says that it also stimulates as a rapid block of the batrachotoxin (BTX) - activated sodium channels of the heart muscle and as an antagonist to long-gating closures.
Literature survey revealed that various potentiometric, high performance liquid chromatographic, capillary zone electrophoresis, spectrofluorimetric, kinetics and pharmacokinetics, spectrometric methods have been developed for the study of PAH [5-19]. Nevertheless, these are considered to be the most selective and sensitive methods that are mainly acknowledged and described well. But on another note, they do have certain disadvantages such as consumption of more time and involvement of tiresome analysis, pricey equipment and great consumption of organic solvents too. Moreover, the pretreatment extraction procedures are being demanded during the analysis of complex matrices including human urine, blood or serum samples [20]. In recent years, usage of different electrochemical methods with various electrodes and modifiers has captured an additional interest of researchers due to speedy analysis, high sensitivity, selectivity, a high response of detection, repeatability and reproducibility, and also are less pricy. Only three reports [21-23] are found in the electrochemical study of PAH till date, amongst which one is from our research contribution itself. As per our literature survey, we found that there is no information regarding the electro-catalytic determination of PAH at the gold electrode. Considering this we here report the usage of the gold electrode for the electrocatalytic behavioral study and determination of PAH in phosphate buffer solution (PBS) and in the presence of different interferences as well. The differential pulse voltammetric (DPV) technique was used to examine the detection limit and quantification limit. The method was successfully applied in demonstrating the determination of PAH in the human biological sample. It was observed that, this method has advantages such as no time consuming sample preparation step prior to drug assay, rapid response, low detection limit, low cost materials and also application of the method in the presence of various excipients makes this electrode material suitable for determination of an analyte even in trace amount.
2. Experimental
2.1. Instrumentation and Analytical Procedure
CHI 630D electrochemical analyser (CH Instruments Inc., of USA) which was coupled with a system of 10 ml single compartment three-electrode glass cell was employed to carry out the electrochemical measurements. A 2 mm diameter gold electrode was used as a primary working electrode (Part No. CHI101), Silver/Silverchloride as a reference electrode and platinum wire as a counter electrode. All the potential measurements were taken against Ag/AgCl (with 3 M KCl solution), Elico LI120 pH meter (of Elico Ltd., India) was used to measure pH. Eppendorf Centrifuge 5804 R was used during urine analysis. An ambient temperature of 25 ┬▒ 0.1┬░C was maintained throughout the experiment.
Prior to use, the polishing of gold electrode has to be done on micro cloths of Buehler that are glued to flat mirrors. A different micro cloth was used every time for each size of the alumina. 0.3, 0.1 and 0.05 ╬╝m were the particle sizes used. The final particle size was 0.05 ╬╝m. After initial cleaning of the electrode, it was merely necessary to polish with 0.05 ╬╝m particle size during the time of experimentation. Before inserting the electrode into the solution, it was made sure to wash and rinse the electrode every time using doubly distilled water. Once this mechanical treatment was done, then the gold electrode was immersed in 0.2 M PBS and various voltammograms were recorded until a stable steady state baseline voltammogram was achieved.
2.2. Reagents and Chemicals
PAH was purchased from Sigma Chemical Company, St. Louis, USA and was used without any purification. A stock solution equivalent to 0.01 M (M = mol/dmŌłÆ3) was prepared using doubly distilled water. According to Christian and PurdyŌĆÖs method [24], supporting electrolyte solution with different pH values varying from 3.0 to 10.4 with a 0.2 M concentration were prepared by adding the suitable amount of sodium hydrogen phosphate, sodium dihydrogen phosphate and trisodium phosphate which is frequently known as phosphate buffer saline (PBS). Other chemical reagents which were used were of analytical/reagent grade, and doubly distilled water was used throughout the experimentation.
2.3. Surface Area of the electrode
A common benchmark redox system, Fe(CN)63ŌłÆ/4ŌłÆ was chosen to determine the electrochemical active surface area of the gold electrode (GE) by using a cyclic voltammetric method and Randles-Sevcik equation. 0.1 M KCl as a supporting electrolyte and 1.0 mM K3Fe(CN)6 as a probe were used to record cyclic voltammograms at different scan rates. Due to the presence of ferricyanide, a well-defined ferricyanide/ferrocyanide redox couple was observed [25]. For a reversible process, the equation is given as follows,
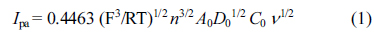
where, Ipa = anodic peak current, n = number of electrons transferred, A0 = surface area of the electrode, D0 = diffusion coefficient, ╬Į = scan rate and C0 = concentration of K3Fe(CN)6 respectively. For 1.0 mM K3Fe(CN)6, T is taken as 298 K, R is 8.314 JKŌłÆ1molŌłÆ1, F is 96,480 C molŌłÆ1, n is 1 and D0 is 7.6 ├Ś 10ŌłÆ6 cm2sŌłÆ1. With the slope of Ipa versus ╬Į1/2 plot, the electroactive surface area of the electrode was calculated. In this experiment we obtained a slope of 2 ├Ś 10ŌłÆ5 ╬╝A(V sŌłÆ1)1/2 and the surface area of the electrode was calculated to be 0.2696 cm2.
2.4. Sample Preparation
Urine samples were collected from a healthy volunteer and were subjected to 5 min centrifugation at 7000 rpm at room temperature (25┬▒0.1┬░C). Then, these samples underwent two-fold dilution with phosphate buffer of pH=7.0 and the test solution were prepared by adding an appropriate amount of standard drug solution to it. The resulting solution was then directly analyzed based on our proposed procedure. The DPV method was then applied for quantifying the drug in human urine samples.
3. Results and Discussion
3.1. Electrochemical Behavior of PAH
In order to evaluate the reaction of PAH at the gold electrode, the electrochemical study was performed employing cyclic voltammetric technique (CV) at pH=7.0. The results are displayed in Fig. 1. It showed no apparent cyclic voltammetric signal in the phosphate buffer solution at a bare gold electrode (curve a). However, an addition of 1.0 mM PAH exhibited two anodic peaks, one at 0.7180 V with an anodic current of 3.0 ┬ĄA and another at 0.9617 V with an anodic current of 15.1 ┬ĄA (curve b) at 50 mVsŌłÆ1scan rate. The cathodic peak that appeared was resultant of the reduction reaction of gold oxides [26].
Fig.┬Ā1.
Cyclic voltammograms at the gold electrode (GE) recorded at scan rate 50 mVsŌłÆ1 in 0.2 M phosphate buffer solution (pH = 7.0): (a) blank run and (b) in presence of PAH 1.0 mM showing sharp oxidation peak.

On the other hand, successive cyclic voltammograms were recorded in which it was seen that the oxidation peak current of PAH shown a noteworthy decrease (Fig. 2). Following first sweep the peak current went on decreasing significantly and finally it was found to remain unchanged. This phenomenon may be recognized due to the fact that the surface of the electrode might be fouled by the adsorption of PAH or by the oxidized product of PAH. Therefore, the voltammograms analogous to the first cycle, that too with the intensified second peak which was observed at 0.9617 V was considered for further experiments in contrary to the broad and blunt first peak.
Fig.┬Ā2.
Successive cyclic voltammograms of 1.0 mM PAH at the gold electrode showing a noteworthy decrease in the oxidation peak current of PAH with increasing cycles. Other conditions are as mentioned in Fig. 1.

3.2. Influence of Supporting Electrolyte
An important stage in the study was to choose an appropriate pH of the supporting electrolyte as the study of the voltammetric behavior of a particular analyte at various pH plays a vital role in the thoughtful understanding of the reaction speed, mechanism and involvement of protons. Hence, the electro-oxidation of 1.0 mM PAH was studied over the pH window of 3.0-10.4 in phosphate buffer solution using cyclic voltammetry. A sharp and well-defined oxidation peak was seen in between pH = 3.0 and pH = 10.4 (Fig. 3). With increase in the pH, the peak potential shifted towards the lesser positive values linearly and the linear relationship between Ep and pH (Fig. 3(a) Inset) can be expressed as shown below,
Fig.┬Ā3.
Influence of pH on the shape of the anodic peak at different pH: 3.0 (a), 4.2 (b), 5.0 (c), 6.0 (d), 7.0 (e), 8.0 (f), 9.2 (g), 10.4 (h), showing highest oxidation peak at pH=7.0. Inset: Plot showing (a) Influence of pH on the peak potential of PAH, (b) Influence of pH on the peak current of PAH. Other conditions are as mentioned in Fig. 1.

The slope of the above equation was found to be 30.0 mV/ pH, which is in agreement with the literature value of 30 mV/pH signifying that two electrons and a proton transfer are participating in the ratedetermining step [27].
It was observed that the peak current was also significantly affected by the solution pH. The peak with highest analytical signal was observed at pH = 7.0 PBS with well-defined anodic voltammetric peak and low background current. Hence, pH = 7.0 was selected as optimum for further voltammetric experimentation (Fig. 3 (b) Inset).
3.3. Influence of Scan Rate
The impact of scan rate study plays an important task in understanding the reaction mechanism and also about the type of the electrode process that was involved. By this study it was possible to acquire the useful information with respect to the electrochemical reaction mechanism from the relationship between peak current and scan rate. Thus, the voltammograms of PAH were recorded at GE surface by varying scan rate using cyclic voltammetry (Fig. 4 A) and linear sweep voltammetry (Fig. 5) at physiological pH = 7.0. Study of an influence of scan rate was done to evaluate the rate limiting step on the gold electrode (diffusion or adsorption-controlled). Ip was proportional to Žģ1/2 in the range from 10-350 mVsŌłÆ1 with the linear relationship for both CV (Fig. 4 B) and LSV which justifies that in the present study, the electrochemical process is of a distinct diffusion controlled [28], with the equation being,
Fig.┬Ā4.
(A) Cyclic voltammograms of 1.0 mM PAH at the gold electrode with different scan rates. (a) -(j) being 10, 25, 50, 75, 100, 150, 200, 250, 300 and 350 mVsŌłÆ1, respectively. Other conditions are as in Fig. 1. (B) Plot of the dependence of oxidation peak current on the square root of scan rate. (C) Plot of the dependence of the logarithm of peak current on logarithm of scan rate. (D) Plot showing the relationship between peak potential and logarithm of scan rate.
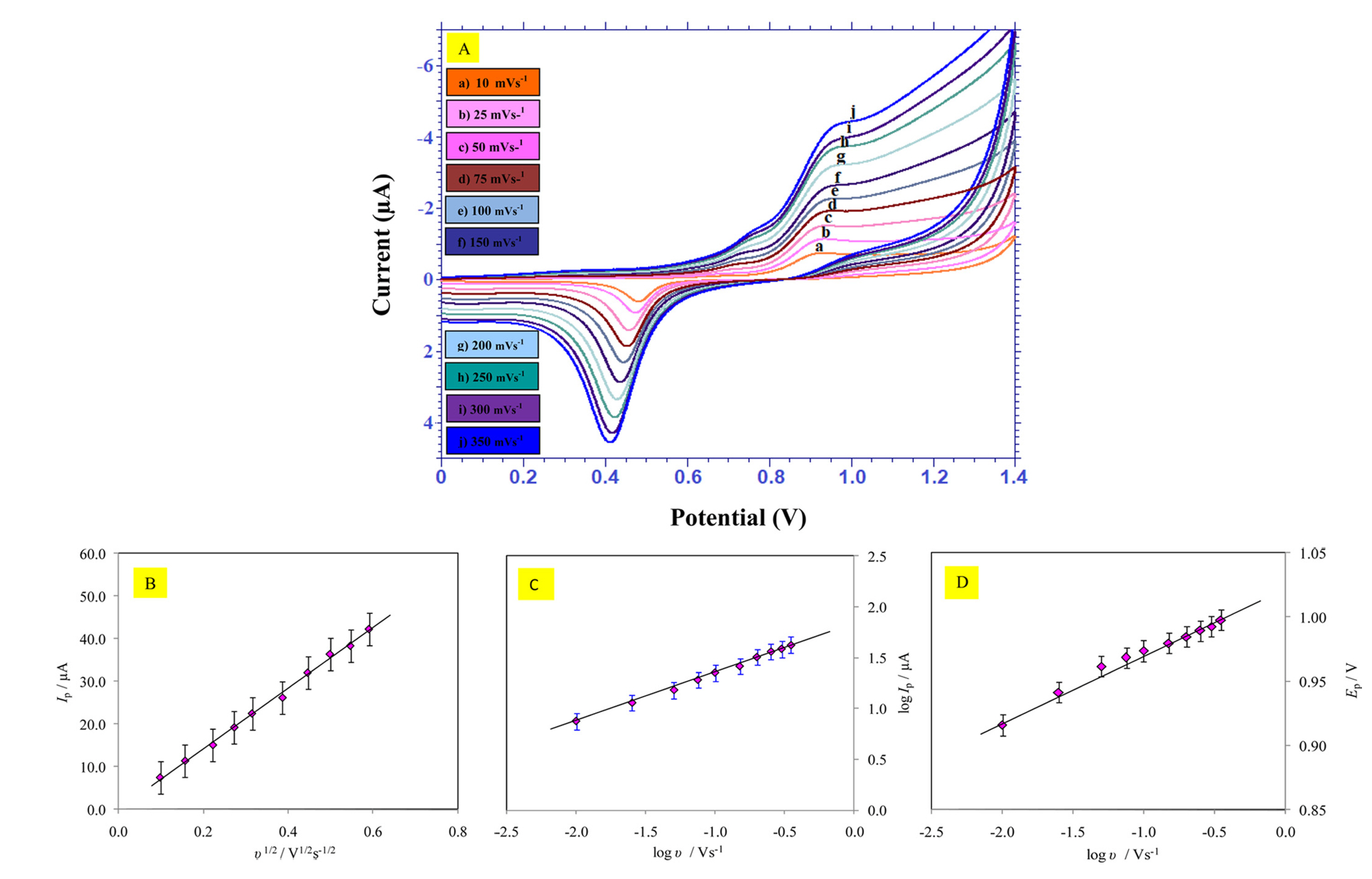
Fig.┬Ā5.
Linear sweep voltammograms of 1.0 mM PAH at the gold electrode with different scan rates. (a)ŌĆō(k) being 10, 25, 50, 75, 100, 150, 200, 250, 300, 350 and 400 mVsŌłÆ1, respectively. Other conditions are as mentioned in Fig. 1. Inset: Plot of the dependence of the logarithm of peak current on logarithm of scan rate.

Further, the plot of logarithm of Ip vs. logarithm of ╬Į showed a straight line with the slope of 0.491 for CV (Fig. 4 C) and 0.547 for LSV (Fig. 5 Inset), which is in concordant to the theoretical literature value of 0.5, a characteristic parameter for a purely diffusion controlled process [28] in which the electroactive species ŌĆō PAH, diffuses from the bulk solution to the planar electrode surface and the equations being expressed as,
We observed that in the presence of PAH, no redox peak was observed during the cathodic run within the studied potential range. This behavior suggests that the process on GE surface is irreversible. Additionally, Ep of the oxidation peak of PAH was also dependent on the ╬Į as Ep was observed to move towards higher positive values with increase in ╬Į values. This behavior substantiates that the reaction at the gold electrode is irreversible. A good linear relationship was observed between peak potential and a logarithm of scan rate for CV (Fig. 4 D) and for LSV which are expressed in the below equations,
For an electrode process which is irreversible, Laviron [29], proposed the relationship between Ep and Žģ which can be defined as,
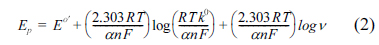
where ╬▒ (alpha) refers to the transfer coefficient, k0 refers to the standard heterogeneous rate constant, n the number of electrons transferred, ╬Į (nu) the scan rate and E0ŌĆÖ refers to the formal redox potential. And other symbols hold their regular meanings. Thus, the slope of Ep vs. logŽģ helps in calculating the value of ╬▒n. In this system, the slope value of 0.050 for CV and 0.047 for LSV were obtained. Substituting the values of R and F at T = 298 K, ╬▒n was calculated to be 1.182 for CV and 1.258 for LSV. Generally, ╬▒ is assumed to be 0.5 in total irreversible electrode process [30]. Further, in both CV and LSV the number of electrons transferred (n) in the electro-oxidation of PAH was calculated to be ~2. In this experiment, we observed that with both CV and LSV techniques analogous results were obtained with respect to the variation of scan rate.
4. Analytical Applications
4.1. Calibration Curve and Detection Limit
As the DPV technique is considered to be the more sensitive technique in comparison to the cyclic voltammetry, DPV was employed further for the quantitative determination of the drug at pH = 7.0. Under the optimized conditions, the plot of peak current vs. concentration of PAH resulted in construction of the calibration curve. The Ip values increased proportionally with the increase in PAH concentration in the linear dynamic range of 0.5 to 30.0 ╬╝M (Fig. 6).
Fig.┬Ā6.
Differential-pulse voltammograms of PAH using the gold electrode recorded at pH = 7.0 phosphate buffer solution at different concentrations: 0.5 (a), 2.5 (b), 7.5 (c), 10.0 (d), 15.0 (e), 20.0 (f), 30.0 (g) ╬╝M. Inset: Plot of the peak current against the concentration of PAH indicating that the Ip values increased proportionally with increase in the PAH concentration.
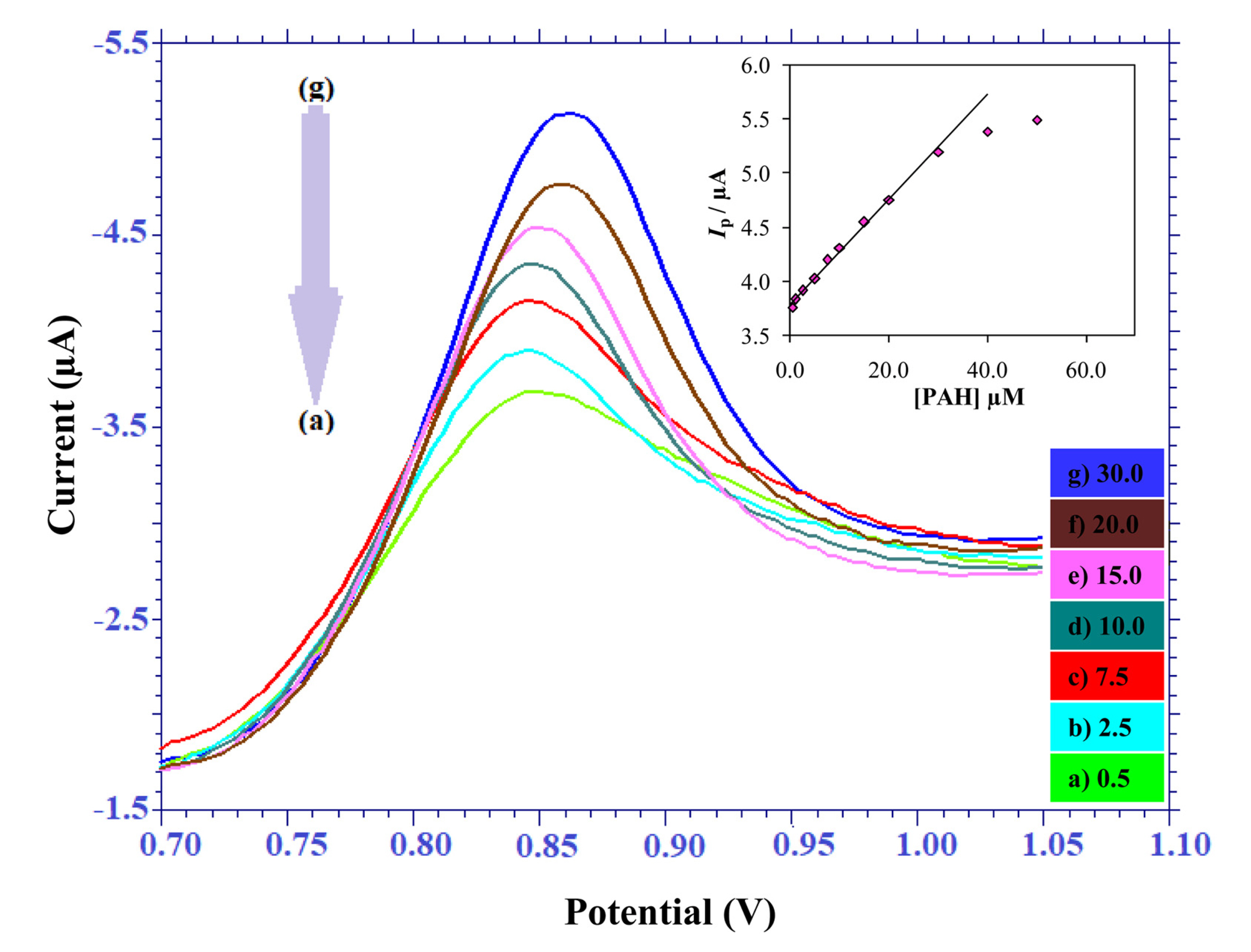
Equation according to the calibration graph is given below,
For more concentrated solution we observed that there was a deviation from linearity in the calibration graph, which can be attributed due to the adsorption of PAH or its oxidised product on the surface of the electrode. In order to develop the relative statistical data of the calibration curve, we carried out five successive determinations.
Using the following equations,
LOD and LOQ were calculated and were found to be 56.4 nM and 18.8 ├Ś 10ŌłÆ8 M, respectively. In the above equation, LOD refers to the limit of detection and LOQ refers to the limit of quantification, s refers to the standard deviation of the peak currents of the blank (five runs), and m refers to the slope of the calibration curve. Table 1 summarizes the limit of detection values reported for some of the classical methods and electrodes and we found that, the present work was better compared to that of the previously reported classical/analytical methods [5,6,11,13,14,19,23].

The precision of the method by intra- and inter-day determination of PAH at two different concentrations (n = 3) was studied within the range of linearity. Table 2 shows the accuracy of the methods expressed as bias% and RSD% for intra and inter days indicating that the proposed method has a high degree of precision.
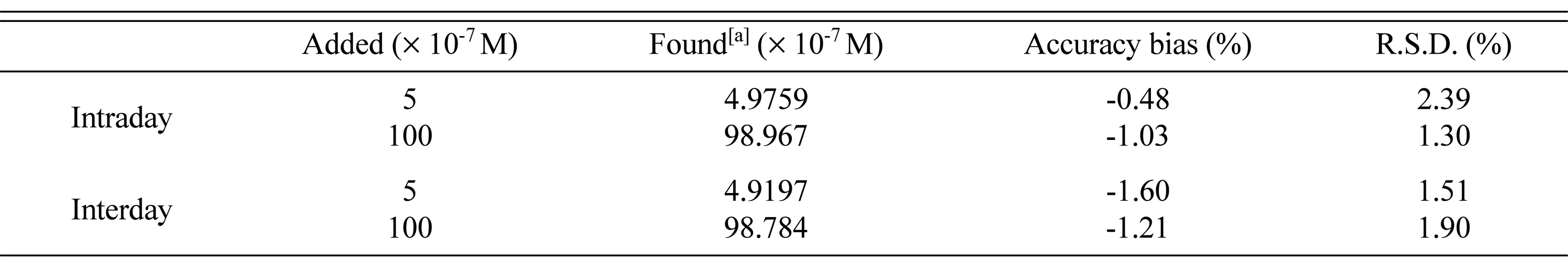
In order to discover the repeatability of the analysis, 6 measurements of 1 ├Ś 10ŌłÆ5 M PAH solution was carried using gold electrode at intervals of 30 min. 2.21% was obtained as the RSD value of peak current which indicated that electrode has a very good repeatability. And coming to the reproducibility between the days, it was same as that of within a day repeatability, with a condition of maintaining unchanged temperature.
4.2. Interference Study
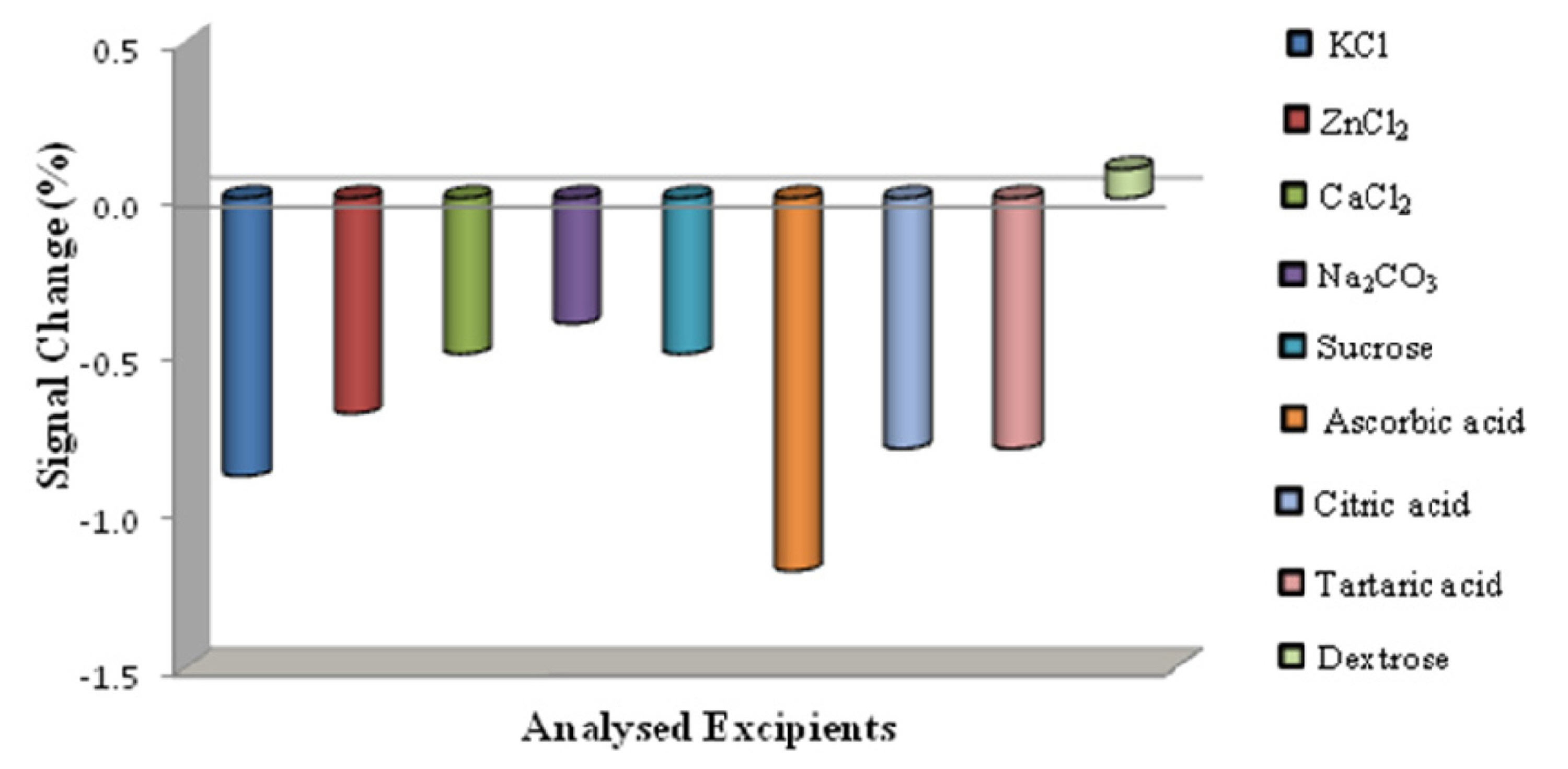
It is well known that potential interfering agents coexist with biological samples, therefore we studied the selectivity of the proposed method by investigating the impact of potential interfering agents coexisting with PAH in biological samples. Approximately ┬▒5% relative error was assumed to be the tolerance limit, when the maximum concentration of the foreign substance was determined. DPV experiments for 1 ├Ś 10ŌłÆ5 M PAH solution in the presence of 1.0 mM of each of the below mentioned excipients was carried out. Fig. 7 displays the experimental results suggesting that, the presence of hundred-fold excess of K+1, Zn+2, Ca+2, Na+1, Sucrose, Ascorbic acid, Citric acid, Tartaric acid and Dextrose changed the potential of the drug slightly but it did not exceed ┬▒5%, indicating that the PAH reactions at the sensing base did not have any effect on the existence of any other metabolites that we tested. Thus, the proposed method can become a selective method.
4.3. Determination of PAH in Spiked Samples of Human Urine
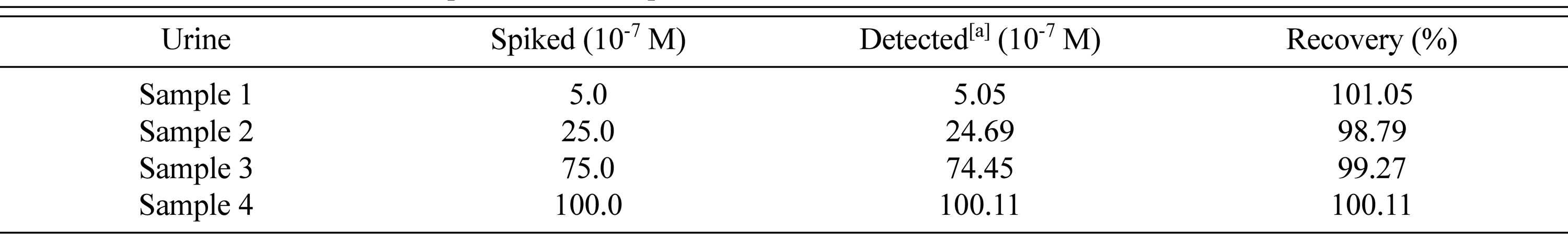
In pharmacokinetic parameter evaluation, usually biological fluids including urine are most commonly analyzed. Sometimes the drug moiety intake, works in the body metabolism completely. But sometimes it so happens that, after responding to the target area or work, it gets excreted from the body. So evaluation of the amount of drug that is excreted becomes very important to know the electrochemical quantification of an analyte. The sample preparation for this analysis was followed as already discussed in subsection 2.4. For determining the spiked PAH concentration in the collected urine samples, the calibration curve was used. Table 3 demonstrates the recoveries obtained from four samples of urine in the percentage range from 98.79 to 101.05 and the R.S.D. was 0.99%.
5. Conclusions
The gold electrode was employed for the first time in studying the electrochemical behavior of PAH in phosphate buffer solution at pH = 7.0. The electrochemical oxidation mechanism of PAH involves transfer of two electrons and a proton and is a typical diffusion-controlled process. Under the selected conditions, it was observed that the peak current was linear to PAH concentrations until a certain range. This helps in voltammetric determination of the analyte as low as 56.4 nM. Besides the high percentage recovery, the study of excipients illustrated that the method was free from the interferences of the commonly used excipients. This method is advantageous as it involve less time consumption, accuracy, as well as the use of simple reagents and apparatus. In addition, this method demonstrates the application of the proposed method as a substitute in real sample clinical trials.




