Cobalt Oxide Nanorods Prepared by a Template-Free Method for Lithium Battery Application
Article information
Abstract
Transition metal oxide-based electrodes for lithium ion batteries have recently attracted much attention because of their high theoretical capacity. Here we report the electrochemical behavior of cobalt oxide nanorods as anodes, prepared by a template-free, one-step electrochemical deposition of cobalt nanorods, followed by an oxidation process. The as-deposited cobalt has a slightly convex columnar structure, and controlled thermal oxidation produces cobalt oxides of different Co/O ratios, while the original shape is largely preserved. As an anode in a rechargeable lithium battery, the Co/O ratio has a strong effect on initial capacity and cycling stability. In particular, the one-dimensional Co@CoxOy core shell structure obtained from a mild heat-treatment results in superior cycling stability.
1. Introduction
Following the report on the electrochemical reaction of transition metal oxides with lithium [1], much research has been pursued to investigate the possible application of these oxides as anode materials for rechargeable lithium batteries [2,3]. Among many transition metal oxides reported, cobalt oxide has drawn special attention owing to its large specific capacity (~900 mAh/g). However, this material might not be suitable for practical use because of the large capacity fade associated with the formation of an electrically insulating product (i.e., lithia), and a thick solid electrolyte interface (SEI) layer. The SEI layer usually reaches several hundreds of nanometers in thickness and readily fills up the internal space of the powder-type porous electrode [1], thereby significantly exacerbating concentration polarization and diminishing the rate capability. In this regard, reports on the excellent cycling stability of mesoporous cobalt oxide nanowire arrays [4] and highly porous nickel oxide networks [5] are worthy of note. A widely open electrode structure not only relieves the lithiation/delithiation-induced stress, but also provides sufficient internal space for rapid transport of ionic species (i.e., lithium ions), despite the formation of the SEI layer during cycling.
The one-dimensional (1-D) nanorod structure has long been considered a promising option for electrodes in rechargeable lithium batteries, particularly when the active materials experience large volume change during operation. Template-assisted methods using porous alumina or polycarbonate are typically used to fabricate 1-D structures [6,7]. However, the use of a template makes the synthetic procedure complicated, and template removal often leads to nanorod aggregation; moreover, this method is cost-prohibitive because a porous template is usually not reusable. Hence, much effort has been directed to the development of a facile synthesis method for 1-D nanorod structures without using a porous template [4,8-11].
Numerous studies have been carried out to investigate the effect of boric acid on the morphology of the electrodeposited cobalt [12], and on the probability of a surface adsorption process to influence the nature of nucleation and growth of the deposits in the Watts solution [13,14]. However, 1-D growth of electrodeposited cobalt has first been reported in our recent publication [15], where we suggested that careful control of the solution chemistry (esp., boric acid content), reduction current density, and temperature would yield electrodeposited cobalt nanorods without the need for a porous template (refer to Figs. 4 and 6 in ref. [15]). Our research on cobalt nanorods is focused on the clarification of the mechanism of nanorod formation, the factors affecting the macroscopic (e.g., uniformity, density) and microscopic (e.g., diameter, aspect ratio) features of the nanorods, and the practical applications of the fabricated cobalt nanorods. In this report, we focus on making use of the oxidized cobalt nanorods as electrodes in rechargeable lithium batteries.

Galvanostatic charge/discharge curves of cobalt oxide electrodes heat-treated at 400 ℃ for (a) 10, (b) 30, and (c) 60 min. (d) shows the dependence of discharge capacity and coulombic efficiency of each sample on cycle number, together with the results of dense cobalt oxide film electrode.
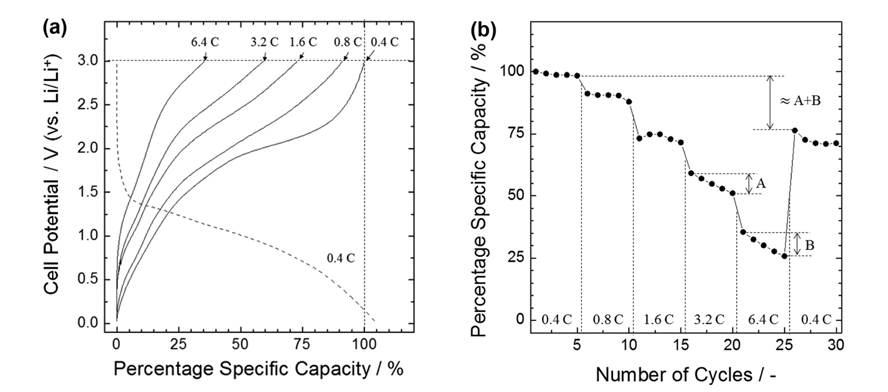
(a) Voltage profiles of cobalt oxide electrode heat-treated at 400℃ for 10 min at different discharging rates. Specific capacity was expressed in percentage relative to discharge capacity at a rate of 0.4 C. (b) Variation of percentage specific capacity with discharging rate.
The cobalt oxide nanorods were prepared by a template-free electrodeposition of cobalt nanorods and subsequent thermal oxidation. The morphology, structure, and composition of the as-prepared cobalt nanorods and the oxidized products were carefully characterized using various techniques. The electrochemical properties were evaluated at different galvanostatic charge-discharge rates for different periods of time, to evaluate the cycling stability and rate performance. Special focus was placed on the effect of the distribution of cobalt and oxygen across the nanorods, and the electrode performance of cobalt@cobalt oxide core-shell nanorods was particularly highlighted.
2. Experimental Section
Stainless steel foil (Alfa Aesar, 0.025 mm thick, Fe:Cr:Ni = 70:19:11) was used as the substrate for the preparation of the electrolytic cobalt nanorod. The stainless steel foil was ultrasonicated for 10 min in a 1:1 volume mixture of ethanol and acetone, washed with distilled water, and then treated with 10 wt% hydrochloric acid to remove the surface residue and native oxide film. A two-electrode electrochemical cell was employed for the electrochemical deposition. The working and counter electrodes were a stainless steel (5 mm × 5 mm) and platinum wire, respectively. The electrolyte was an aqueous solution of 0.05 M CoSO4·7H2O (> 99 %, Aldrich) and 0.4 M H3BO3 (> 99.5 %, Junsei), and the temperature was maintained at 15℃. The cobalt nanorods were electrodeposited for 20 s at a constant current density of 150 mA/cm2 using an EG&G 263A potentiostat/galvanostat. The as-prepared cobalt nanorods were washed with distilled water and dried at room temperature. In order to prepare the cobalt oxides, the asprepared cobalt nanorods were heat-treated in air at 400℃ for 10, 30, or 60 min. For the sake of comparison, a dense cobalt thin film with a thickness of ~400 nm was also electroplated for 10 s at 30 ℃ in an aqueous solution of 1 M CoSO4·7H2O and 2 M H3BO3. The applied current density was 50 mA/cm2. The sample was then heat-treated in air at 400℃ for 10 min to produce a cobalt oxide thin film.
The morphology, chemical composition, and crystal structure of the as-prepared and heat-treated samples were examined with a field-emission scanning electron microscope (FE-SEM, S-4800, Hitachi, Japan), energy-dispersive X-ray spectrometer (EDS, EMAX 7593-H, Horiba, Japan), and X-ray diffractometer (XRD, D8 Advance, Bruker, Germany), respectively. In addition, single nanorods were cut parallel to their central axes using a focused ion beam (FIB, Versa3D LoVac, FEI, USA), and transmission electron microscope (TEM) / EDS analysis (Titan G2 ChemiSTEM Cs Probe, FEI, USA) was carried out to gain an in-depth understanding of the composition of the samples.
For the electrochemical studies of the heat-treated cobalt oxide nanorods as the anode in a rechargeable lithium battery, a three-electrode electrochemical cell was employed. Lithium foil was used as the counter and reference electrodes. The electrolyte was a 1 M solution of lithium hexafluorophosphate (LiPF6) in a 1:1 volume mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC). The cells were galvanostatically cycled at a rate of 0.4 C from 0.01 to 3.0 V vs. Li/Li+ (C rate was calculated on the basis of the experimentally measured reversible capacity). To estimate specific capacity, the measured capacity was divided by film mass, which was determined by subtracting the substrate mass from the total (film + substrate) mass. To evaluate the rate capability, the cell was first charged to 0.01 V vs. Li/Li+ at a rate of 0.4 C, and then discharged to 3.0 V vs. Li/Li+ at rates ranging from 0.4 to 6.4 C. A BaSyTec CTS-Lab battery tester was used for the galvanostatic charge/discharge experiments. All the cells were assembled and tested in a glove box (MBraun, Germany) filled with purified argon gas.
3. Results and Discussion
Figs. 1a-c show the morphology of the cobalt that was electrodeposited on the stainless steel substrate at a current density of 150 mA/cm2 in an aqueous solution of 0.05 M CoSO4·7H2O and 0.4 M H3BO3. The electrodeposited cobalt has a slightly convex 1-D columnar or nanorod shape with a diameter of 200-400 nm, and was uniformly distributed through the substrate. For use in the anode of a rechargeable lithium battery, the as-prepared cobalt nanorods were heat-treated in air to form cobalt oxide nanorods. The rod diameter increased (400-800 nm) after heat-treatment for 10 min, but the overall integrity of the sample was maintained in terms of nanorod shape and open structure (Fig. 1d). The images of the samples after heat-treatment for 30 and 60 min are not shown, since the structural features are essentially the same as those of the sample heat-treated for 10 min.

(a) Top, (b) cross sectional, and (c) inclined views of the as-deposited cobalt on a stainless steel substrate; (d) the inclined view of the sample that was heat-treated at 400 ℃ for 10 min.
In Fig. 2a the compositional analysis of the as-prepared electrodeposited cobalt is shown. Apart from the stainless steel substrate (i.e., Fe, Ni, Cr) signals, the only remaining meaningful signal could be assigned to metallic cobalt, indicating that the nanorods were pure cobalt. Structural analysis revealed that when the cobalt was heat-treated at 400 ℃, it was transformed to cobalt oxide, Co3O4, having a cubic spinel structure (Fig. 2b). As the duration of heat-treatment increased, the Co3O4 phase peaks intensified and sharpened, indicating improved crystallinity.
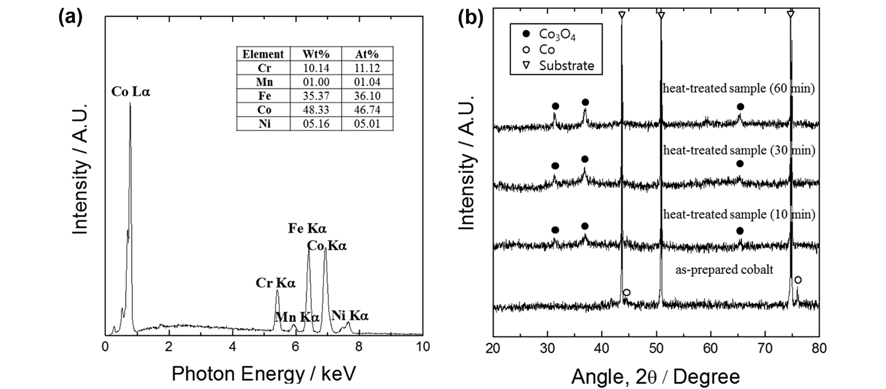
(a) EDS result of the as-deposited cobalt, and (b) XRD patterns of the as-deposited and heat-treated samples.
Fig. 3 shows the compositional depth profiles of heat-treated single nanorods, obtained from a combination of FIB and TEM/EDS analyses. Notably, there were two different regions of pure cobalt core and cobalt oxide surface layer in the sample heat-treated for 10 min (Fig. 3a), indicating that the inter-diffusion of cobalt and oxygen was an ongoing process, but the oxygen did not reach the central area of the nanorod. The average ratio of Co to O in cobalt oxide surface layer was 0.69:0.31. Considering that the structural analysis showed that the Co3O4 spinel was the only crystalline phase in the sample (Fig. 2b), this ratio suggests that the sample consisted of a combination of the Co3O4 phase and non-stoichiometric compounds CoOd, where d < 1. By simply writing the Co3O4/CoOd composite as CoxOy, the above unique structure can be denoted as a 1-D Co@CoxOy core shell nanostructure.

Electron microscope images of single nanorods that were cut parallel to their central axes using a focused ion beam. Their EDS compositional depth profiles are superimposed on the images. (a), (b), and (c): samples heat-treated at 400 ℃ for 10, 30, and 60 min, respectively.
In the case of the sample heat-treated for 30 min (Fig. 3b), in spite of slight fluctuation of Co and O contents, they were relatively uniformly distributed across the sample. The ratio of Co to O was averaged to be 0.53:0.47. Again, the Co3O4 spinel was the only crystalline phase in the sample (Fig. 2b), and this sample could also be denoted as a composite of the Co3O4 spinel and amorphous CoOd (d<1). As the heat-treatment duration was increased to 60 min, the Co:O ratio came close to 3:4 (Fig. 3c), indicating that the non-stoichiometric compounds were transformed to the Co3O4 phase.
Figs. 4a, 4b, and 4c present the galvanostatic charge/discharge curves, obtained from the samples heat-treated for 10, 30, and 60 min, respectively. For the first charging process, the large irreversible capacity loss was most likely due to the formation of a solid electrolyte interphase (SEI) layer [16]. In the subsequent cycles, the sample reacted quite reversibly with lithium. The variations in the specific capacity and coulombic efficiency of the samples with the cycle number are summarized in Fig. 4d. The initial specific capacity increased as duration of heat-treatment increased since oxygen content in the sample became higher. All the three samples showed almost no capacity fade up to ~20 cycles. Notably, after 20 cycles, the sample heat-treated for 10 min (i.e., 1-D Co@Co3O4 core-shell nanostructure) showed the highest capacity retention, whereas when the sample was heat-treated for a longer time (30 or 60 min), the reversible capacity decreased relatively quickly.
These results indicate that there is a trade-off between the specific capacity and cycling stability, and that this is possibly caused by the oxygen content in the samples (the Co:O ratios of samples heat-treated for 10, 30, and 60 min, were 0.69:0.31, 0.53:0.47, and 0.43:0.57, respectively). It is conceivable that the higher oxygen content enables the production of a larger amount of reversible lithia (Li2O) in the charging process, and is beneficial to specific capacity. However, the higher oxygen content also causes the sample to degrade upon cycling, due to the increase in formation of lithia and SEI layers, which are known to promote the deterioration of active materials by increasing the inter-nanograin distance of the metallic phase in it [16,17]. Nevertheless, the specific reversible capacities in all three samples tested in this work were outstanding, compared to that of the dense Co3O4 film electrode, and exceeded the theoretical capacity of graphite even after 50 cycles.
In the dense Co3O4 film prepared by the thermal oxidation of a 400 nm-thick cobalt film at 400℃ for 10 min, the reversible capacity decreased very quickly with cycling, and fell below 30% of the initial capacity after 10 cycles (Fig. 4d); these results are consistent with previous reports [18]. When the nanorod sample heat-treated for 10 min was chosen for the comparison, apart from some exceptional data of graphene-based materials [19,20], its discharge capacity retention (~71 % at 50th cycle with respect to initial capacity) was comparable to those obtained from novel structures like mesoporous Co3O4 nanowire arrays [4], and Co3O4/graphene hybrid anode [21]. Since the sample has a wide range of discharge (delithiation) potential window and most of its discharge capacity is obtained in a relatively high potential range between 1.0 and 2.0 V vs. Li/Li+, high-voltage cathode materials with a potential over 4.0 V vs. Li/Li+ such as LiMexPO4 (Me = Mn, Ni, Co, and V) [22-25] and LiNi0.5Mn1.5O4 [26] can be used against the anode sample suggested in this work for full cell construction.
Fig. 5 shows the surface morphology of the samples after 50 charge-discharge cycles. A comparison of the images of the sample heat-treated for 10 min, before (Fig. 1d) and after (Fig. 5a) cycling revealed that, while the long nanorods were seen after the cycling even though their diameter was enlarged to a few micrometers, most of the short nanorods looked a little bit flattened. It is probable that the SEI layer filled up the inter-nanorod space during cycling and hence the short nanorods were partially buried. The change in shape after cycling became more pronounced for the samples that had undergone longer heat-treatment, due to their higher oxygen content, resulting in a more vigorous SEI layer formation. This implies that the cycling-induced structural change and performance degradation of the nanorods prepared in this work are unavoidable. Further studies are needed, to preserve the original structure during prolonged cycling.

Electron microscope images of the samples heat-treated at 400℃ for (a) 10, (b) 30, and (c) 60 min after 50 chargedischarge cycles.
The rate capability was evaluated for the sample heat-treated for 10 min, which showed the highest cycling stability and the least shape change. The voltage profiles obtained at different discharging rates, and a summary of the rate performance, are given in Figs. 6a and 6b, respectively. At discharging rates of 1.6 C and 6.4 C, the sample retained about 75 % and 30 % of the capacity at a rate of 0.4 C, respectively. While the rate capability appeared to be moderate, it cannot actually be compared to those in the literature, due to its strong dependence on cell design. That is, a change in bulk structure and surface of the active materials, electrolyte chemistry, separator (if any), location of working and counter electrodes, etc., might lead to different results. Nevertheless, the capacity retention at relatively high discharging rates is noteworthy. The electrode appeared to degrade quickly while discharging at the rates of 3.2 and 6.4 C (designated as “A” and “B” in the figure, respectively). This makes a difference in the specific capacities at a rate of 0.4 C, measured before and after the rate performance test (“≈A+B” in the figure). The reason for the larger capacity fade at higher C rate is yet to be determined, and we found no consensus on the rate dependence of capacity retention in literatures [4,19-21,27]. Since it is known that the growth of SEI layer and the resultant increase in the inter-nanograin distance of metals in active materials are possible causes of poor cycling stability of transition metal oxide anodes [16], further work needs to focus on the effect of high current drain on the formation and chemistry of SEI, together with the microstructural changes in active materials after the SEI formation.
4. Conclusions
(1) Cobalt nanorods with diameters of 200-400 nm were successfully synthesized by an electrochemical deposition process. Upon thermal oxidation, the cobalt nanorods were converted to cobalt oxide nanorods but the shape was essentially preserved. The oxide had a cubic spinel structure and the crystallinity increased with the duration of heat-treatment.
(2) Detailed structural and compositional analyses suggest that the cobalt oxides in the samples heat-treated for 10 and 30 min consisted of the spinel Co3O4 and some amorphous CoOd (d<1) phases, while the sample heat-treated for 60 min had a pure spinel Co3O4 phase. In particular, the shallow interdiffusion between Co and O due to mild heat-treatment for 10 min resulted in a 1-D Co@CoxOy core shell nanostructure.
(3) The cobalt oxide nanorods reacted reversibly with lithium when used as the anode in a rechargeable lithium battery. A shorter duration of heat-treatment resulted in a lower specific capacity but a higher capacity retention. It appears that the oxygen content in the nanorods is responsible for the trade-off between specific capacity and cycling stability.
(4) The 1-D Co@CoxOy core shell nanostructure showed excellent capacity retention (almost no capacity loss over 20 cycles; ~71 % at 50th cycle with respect to initial capacity) at a rate of 0.4 C. However, higher current densities (greater than ~3 C rate) degraded the sample very quickly. Further studies are necessary to gain a profound understanding of SEI formation and microstructural change of active material under the conditions of high-rate operation.
Acknowledgements
This work was supported by the Financial Supporting Project of Long-term Overseas Dispatch of PNU’s Tenure-track Faculty, 2015.
References
P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J-M. Tarascon, Nature, 2000, 407,496-499.
Poizot P., Laruelle S., Grugeon S., Dupont L., Tarascon J-M.. Nature 2000;407:496–499. 10.1038/35035045.D. Larcher, G. Sudant, J-B. Leriche, Y. Chabre and J-M. Tarascon, J. Electrochem. Soc., 2002, 149, A234-A241.
Larcher D., Sudant G., Leriche J-B., Chabre Y., Tarascon J-M.. J. Electrochem. Soc. 2002;149:A234–A241. 10.1149/1.1435358.N. Jayaprakash, W. D. Jones, S. S. Moganty and L. A. Archer, J. Power Sources, 2012, 200, 53-58.
Jayaprakash N., Jones W. D., Moganty S. S., Archer L. A.. J. Power Sources 2012;200:53–58. 10.1016/j.jpowsour.2011.10.018.Y. Li, B. Tan and Y. Wu, Nano Letters, 2008, 8, 265-270.
Li Y., Tan B., Wu Y.. Nano Letters 2008;8:265–270. 10.1021/nl0725906.W.-S. Choi, W. Chang and H.-C. Shin, J. Solid State Electrochem., 2014, 18, 427-433.
Choi W.-S., Chang W., Shin H.-C.. J. Solid State Electrochem. 2014;18:427–433. 10.1007/s10008-013-2272-3.M. Nishizawa, K. Mukai, S. Kuwabata, C. R. Martin and H. Yoneyama, J. Electrochem. Soc., 1997, 144, 1923-1927.
Nishizawa M., Mukai K., Kuwabata S., Martin C. R., Yoneyama H.. J. Electrochem. Soc. 1997;144:1923–1927. 10.1149/1.1837722.A. Azarian, A. Iraji zad, A. Dolati and S. M. Mahdavi, Thin Solid Films, 2009, 517, 1736-1739.
Azarian A., Iraji zad A., Dolati A., Mahdavi S. M.. Thin Solid Films 2009;517:1736–1739. 10.1016/j.tsf.2008.09.057.J. Wang, Y. L. Bunimovich, G. Sui, S. Savvas, J. Wang, Y. Guo, J. R. Heath and H.-R. Tseng, Chem. Comm., 2006, 3075-3077.
Wang J., Bunimovich Y. L., Sui G., Savvas S., Wang J., Guo Y., Heath J. R., Tseng H.-R.. Chem. Comm. 2006;:3075–3077.S. Saadat, Y. Y. Tay, J. Zhu, P. F. Teh, S. Maleksaeedi, M. M. Shahjamali, M. Shakerzadeh, M. Srinivasan, B.Y. Tay, H. H. Hng, J. Ma and Q. Yan, Chem. Mater., 2011, 23(4), 1032-1038.
Saadat S., Tay Y. Y., Zhu J., Teh P. F., Maleksaeedi S., Shahjamali M. M., Shakerzadeh M., Srinivasan M., Tay B.Y., Hng H. H., Ma J., Yan Q.. Chem. Mater. 2011;23(4):1032–1038. 10.1021/cm103068v.S. H. Park, H. S. Shin, Y. H. Kim, H. M. Park and J. Y. Song, Nanoscale, 2013, 5, 1864-1869.
Park S. H., Shin H. S., Kim Y. H., Park H. M., Song J. Y.. Nanoscale 2013;5:1864–1869. 10.1039/c2nr33056c.S. H. Park, H. S. Shin, Y. H. Kim, H. M. Park and J. Y. Song, J. Alloys and Compds., 2013, 580, 152-156.
Park S. H., Shin H. S., Kim Y. H., Park H. M., Song J. Y.. J. Alloys and Compds. 2013;580:152–156. 10.1016/j.jallcom.2013.05.130.R. Sivasubramanian and M. V. Sangaranarayanan, Mater. Chem. Phys., 2012, 136, 448-454.
Sivasubramanian R., Sangaranarayanan M. V.. Mater. Chem. Phys. 2012;136:448–454. 10.1016/j.matchemphys.2012.07.006.J. P. Hoare, J. Electrochem. Soc., 1986, 133, 2491-2494.
Hoare J. P.. J. Electrochem. Soc. 1986;133:2491–2494. 10.1149/1.2108456.J. Horkans, J. Electrochem. Soc., 1979, 126, 1861-1867.
Horkans J.. J. Electrochem. Soc. 1979;126:1861–1867. 10.1149/1.2128816.S.-J. Kim and H.-C. Shin, Kor. J. Mater. Res., 2014, 24(6), 319-325.
Kim S.-J., Shin H.-C.. Kor. J. Mater. Res. 2014;24(6):319–325. 10.3740/MRSK.2014.24.6.319.P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, J. Power Sources, 2001, 97, 235-239.
Poizot P., Laruelle S., Grugeon S., Dupont L., Tarascon J.-M.. J. Power Sources 2001;97:235–239.S. Grugeon, S. Laruelle, R. Herrera-Urbina, L. Dupont, P. Poizot and J. M. Tarascon, J. Electrochem. Soc., 2001, 148, A285-A292.
Grugeon S., Laruelle S., Herrera-Urbina R., Dupont L., Poizot P., Tarascon J. M.. J. Electrochem. Soc. 2001;148:A285–A292. 10.1149/1.1353566.C. Wang, D. Wang, Q. Wang and L. Wang, Electrochim. Acta, 2010, 55, 6420-6425.
Wang C., Wang D., Wang Q., Wang L.. Electrochim. Acta 2010;55:6420–6425. 10.1016/j.electacta.2010.06.044.Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4(6), 3187-3194.
Wu Z.-S., Ren W., Wen L., Gao L., Zhao J., Chen Z., Zhou G., Li F., Cheng H.-M.. ACS Nano 2010;4(6):3187–3194. 10.1021/nn100740x.S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Mullen, ChemSusChem, 2010, 3, 236-239.
Yang S., Cui G., Pang S., Cao Q., Kolb U., Feng X., Maier J., Mullen K.. ChemSusChem. 2010;3:236–239. 10.1002/cssc.200900106.H. Kim, D.-H. Seo, S.-W. Kim, J. Kim and K. Kang, Carbon, 2011, 49, 326-332.
Kim H., Seo D.-H., Kim S.-W., Kim J., Kang K.. Carbon 2011;49:326–332. 10.1016/j.carbon.2010.09.033.D. Choi, D. Wang, I. T. Bae, J. Xiao, Z. Nie, W. Wang, V. V. Viswanathan, Y. J. Lee, J.-G. Zhang, G. L. Graff, Z. Yang and J. Liu, Nano Lett., 2010, 10, 2799-2805.
Choi D., Wang D., Bae I. T., Xiao J., Nie Z., Wang W., Viswanathan V. V., Lee Y. J., Zhang J.-G., Graff G. L., Yang Z., Liu J.. Nano Lett. 2010;10:2799–2805. 10.1021/nl1007085.J. Ni, H. Wang, L. Gao and L. Lu, Electrochim. Acta, 2012, 70, 349-354.
Ni J., Wang H., Gao L., Lu L.. Electrochim. Acta 2012;70:349–354. 10.1016/j.electacta.2012.03.080.X. Rui, X. Zhao, Z. Lu, H. Tan, D. Sim, H. H. Hng, R. Yazami, T. M. Lim and Q. Yan, ACS Nano, 2013, 7, 5637-5646.
Rui X., Zhao X., Lu Z., Tan H., Sim D., Hng H. H., Yazami R., Lim T. M., Yan Q.. ACS Nano 2013;7:5637–5646. 10.1021/nn4022263.M. Choi, H.-S. Kim, Y. M. Lee and B.-S. Jin, J. Electrochem. Sci. Technol., 2014, 5, 109-114.
Choi M., Kim H.-S., Lee Y. M., Jin B.-S.. J. Electrochem. Sci. Technol. 2014;5:109–114. 10.5229/JECST.2014.5.4.109.X. Zhang, F. Cheng, J. Yang and J. Chen, Nano Lett., 2013, 13, 2822-2825.
Zhang X., Cheng F., Yang J., Chen J.. Nano Lett. 2013;13:2822–2825. 10.1021/nl401072x.N. Jayaprakash, W. D. Jones, S. S. Moganty and L. A. Archer, J. Power Sources, 2012, 200, 53-58.
Jayaprakash N., Jones W. D., Moganty S. S., Archer L. A.. J. Power Sources 2012;200:53–58. 10.1016/j.jpowsour.2011.10.018.