1. Introduction
Chlorine (Cl2) and chlorine oxyanions (ClOx−, x = 1–4) play a significant role in over 50% of industrial processes, such as chemical, pharmaceutical, water sanitation, bleaching, and fuel industries [1–3]. In 2018, global production of Cl2 reached 88.9 million tons [3]. Cl2 production relies mainly on chlor–alkali electrolysis. In a chlor–alkali electrolyzer, the chlorine evolution reaction (CER, Equation 1) generates gaseous Cl2 at the anode, which is counterbalanced by the hydrogen evolution reaction (HER) at the cathode (Fig. 1) [1,2,4].
Because the anodic CER requires a larger overpotential than the HER, the design of highly active, selective, and long-term durable CER electrocatalysts is of prime importance in the chlor–alkali process.
Precious metal-based mixed metal oxides (MMOs, e.g., RuO2–TiO2) have been predominantly used as anode electrocatalysts for the CER during the past half-century, with a notable example of the dimensionally stable anode (DSA) [5–8]. MMO catalysts exhibit decade-long stability and superior CER activity even under corrosive conditions [6,7]. However, they also catalyze the acidic oxygen evolution reaction (OER, Equation 2) as a parasitic reaction that competes with CER [7,8].
Comparing the two reactions, OER is thermodynamically favored over CER because of its lower equilibrium potential. However, the CER involving two-electron (2e−) transfer exhibits much faster kinetics than the OER involving 4e− transfer. Therefore, these two reactions are intrinsically competitive. Given that the chlor–alkali process consumes at least 250 TWh of electricity annually [3], a small amount of O2 generation can lead to significant energy loss. Even the chlor–alkali cell operated under OER-suppressing conditions (e.g., highly acidic pH, saturated Cl−, and increased temperature) inevitably generates more than 5% O2 at the anode [8,9]. Moreover, H2 produced from the cathode can cross through the membrane and mix with the O2 byproduct, generating explosive hazards and increasing the cost of additional workup [4,9].
Recently, Cl2 production under neutral pH and low Cl− concentration conditions has become increasingly important in diverse applications [10–13]. As available freshwater has become insufficient, seawater and wastewater have recently been regarded as cost-effective and renewable alternative salines [11–13]. Cl2 generated from alternative saline is currently utilized for the disinfection of ship ballast water using seawater [14,15] and for the electrochemical oxidation (EO) of trace organic chemicals in wastewater [15]. On the other hand, seawater filtration (e.g., reverse osmosis) has become an important technology to supply freshwater to the coastal area [16,17]. However, O2-depleted effluents consist of concentrated Cl− (>1.0 M), and thus their discharge directly into the ocean is prohibited [17,18]. Selective CER can reduce the Cl− concentration of this concentrated effluent to make it disposable and generate useful Cl2 for water treatment [18]. At neutral pH, gaseous Cl2 or Cl− ion is spontaneously hydrolyzed (Equation 3) or electrochemically oxidized (Equation 4) to generate HClO, respectively. The resulting HClO acts as a strong oxidant with a long residual time, which is suitable for disinfection and chemical neutralization [2,8].
The above examples illustrate the increasing importance of Cl2 production in neutral and low Cl− concentration electrolytes. However, the selectivity issue between the CER and OER becomes more prominent under these conditions, and the electrocatalytic efficiency of the CER is much lower than that of the chlor–alkali process [8,9].
Hence, the development of CER-selective electrocatalysts is key to advancing Cl2-generating devices as well as the chlor–alkali process. MMO catalysts primarily consist of precious metals (Ru, Ir, and Pt) as a catalyst layer and group 4 and 5 transition metals (Ti, Ta, Nb, and Zr) as a conductive backbone, and these two metals can easily form a mixed oxide surface [9,19]. Importantly, mixing precious metals with transition metals endows MMOs with long-term stability under highly corrosive and oxidizing operating conditions of CER [7,8]. Several approaches have been exploited to improve the activity and stability of MMOs for the CER, including the doping of foreign metals into the MMO backbone to modify the electronic structure [8,20] and tailoring of MMO nanostructures to increase the electrochemically active surface area (ECSA) and promote bubble removal [21–23]. In addition, new CER catalysts based on non-precious metal oxide compositions [24–27] and atomically dispersed catalysts (ADCs) [28,29] have been explored. In addition to tuning the catalytic materials, the CER selectivity can also be controlled by optimizing the supporting electrolytes [8,30], membranes [4,31], and cell structures [4,31].
This review focuses on the fundamental aspects and catalyst design strategies for the electrocatalysis of selective CER. In Section 2, experimental and theoretical approaches to elucidate the active sites of MMO catalysts and the scaling relationship between the CER and OER inherent in MMOs are summarized. In addition, kinetic analysis methods for understanding the reaction kinetics and mechanisms are presented. In Section 3, new CER catalyst design strategies that can enhance CER activity and selectivity and overcome the scaling relationship are discussed, which includes the use of dopants and the exploitation of non-precious metal catalysts and ADCs.
2. Fundamental Aspects of Selective CER
2.1 Experimental approaches
A structural understanding of the active sites is essential for establishing the structure–property relationship in catalysis. Such insights are particularly critical for metal oxide catalysts, where multiple active sites can emerge. DSA, an archetype of MMO-based CER catalysts, typically consists of rutile RuO2 and rutile TiO2 with optional doping of other precious or transition metals [32]. Notably, DSA preserves its surface structure even after being operated on for over 10 years [33]. Given that rutile RuO2 alone is prone to facile dissolution via ionization under acidic electrochemical operation conditions [33–35], the active sites of DSA with remarkable stability could originate from the interfaces where RuO2 and TiO2 are mixed.
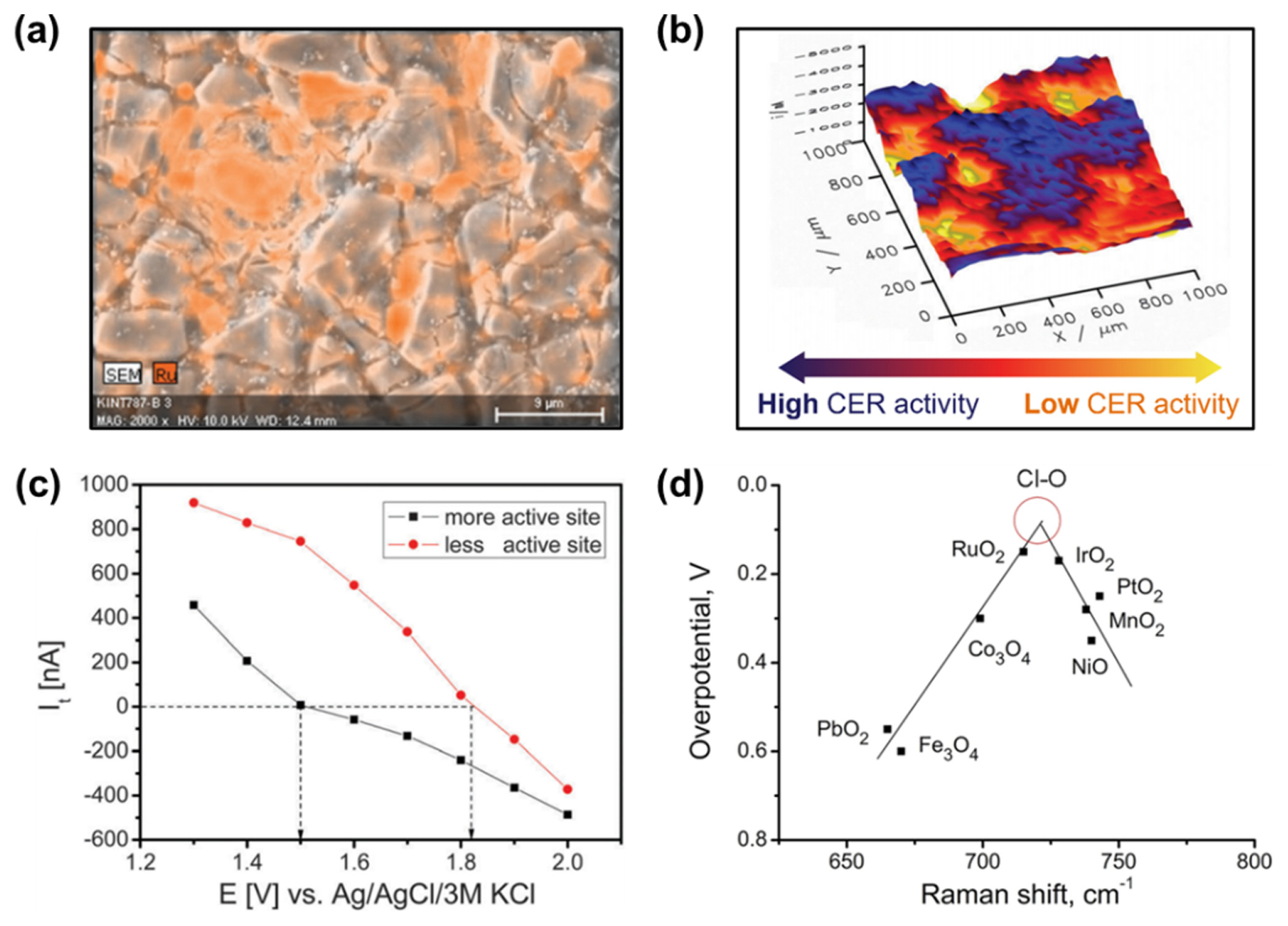
Zeradjanin et al. imaged the surface of a RuO2–IrO2–TiO2-based DSA electrode using scanning electron microscopy–energy dispersive X-ray spectroscopy (SEM–EDS), which showed random cracks and non-uniform distribution of Ru species (Fig. 2a) [36]. They prepared DSA electrodes with controlled crack sizes and correlated their sizes with their CER activities and conductivities [22]. As the channel width in the DSA cracks increased, CER activity increased proportionally. This correlation suggests that a high density of active RuO2–TiO2 interfacial species was distributed on the channels. Moreover, wider channels allowed for the facile detachment of Cl2 bubbles, generating a large available surface area for the CER.
Scanning electrochemical microscopy (SECM) is an analytical method that can visualize the CER process in situ at the micron scale by reducing the generated Cl2 molecules with a scanning tip [37]. Zeradjanin et al. identified from 3D SECM images that different types of active sites existed on the RuO2–IrO2–TiO2 electrode (Fig. 2b). The tip currents near the sample, measured as a function of the applied potential over two different sample regions, clearly showed different catalytic activity trends (Fig. 2c) [36]. Chen et al. revealed using SECM that the area showing high CER activity became wider as the crack channel size in the RuO2–SnO2–TiO2-based electrode increased [38].
In situ Raman spectroscopy can also serve as a versatile tool for analyzing the reaction intermediates adsorbed on catalyst surfaces, thus providing detailed information on the active sites. Zeradjanin et al. revealed the relationship between the Raman shift for Cl–O bonding and the CER overpotential for metal oxide catalysts (Fig. 2d) [39]. RuO2 and IrO2 occupy the near-top position in the volcano-shaped plot with intermediate Raman shifts among the examined compositions. This correlation suggests that the CER proceeds via Cl species adsorbed on the oxygen site rather than on the metal site, which has been verified by theoretical calculations (see below). Importantly, this result suggests that the vibration frequencies of the crystal lattice can serve as dynamic catalytic descriptors instead of thermodynamic descriptors, such as the adsorption energy of intermediates.
2.2 Theoretical approaches
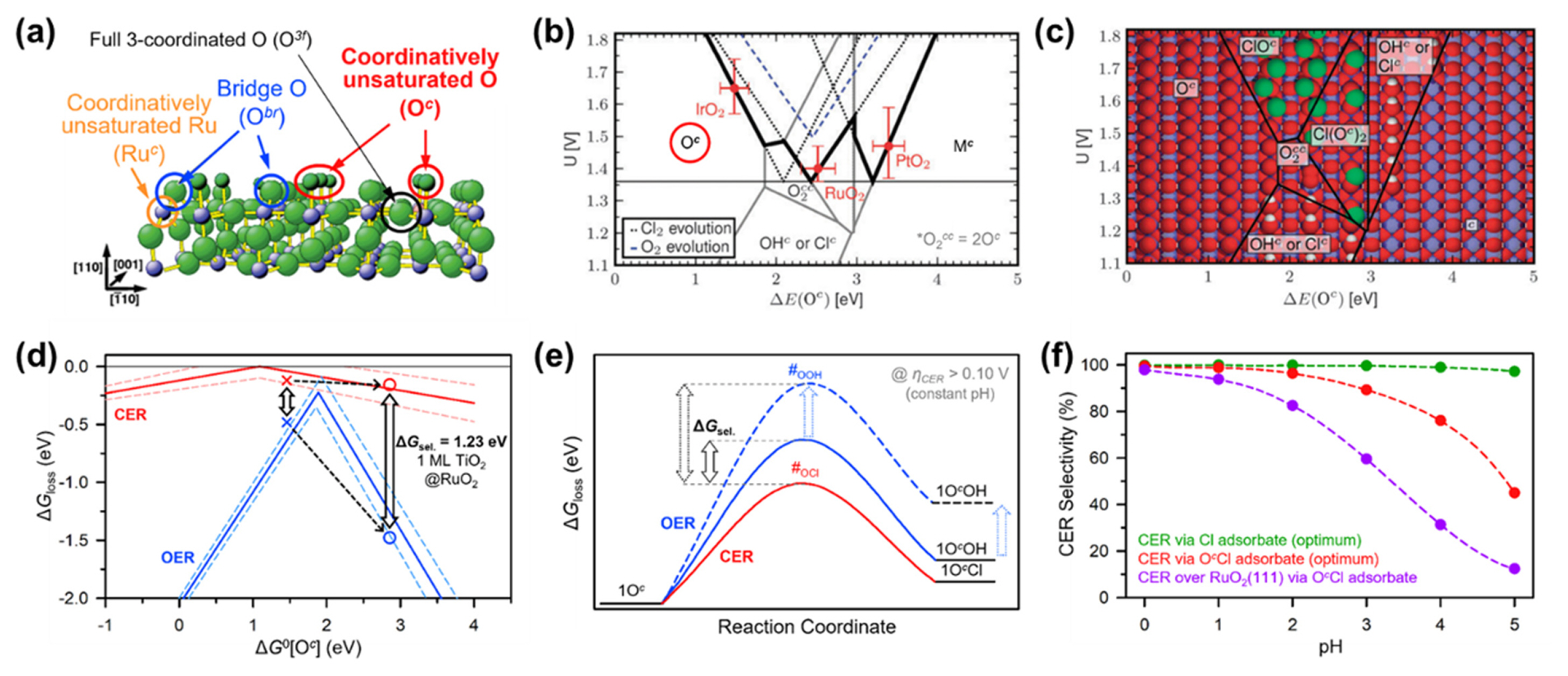
Recently, a growing body of theoretical studies, mainly based on density functional theory (DFT), has provided new insights into the fundamental understanding of CER electrocatalysis [40,41]. DFT calculations have helped to identify the active sites of metal oxide-based CER electrocatalysts and predict the reaction pathways in competitive electrocatalytic reactions between the CER and OER [9,40]. Over et al. proposed possible active sites on the stoichiometric surface of RuO2(110) (Fig. 3a) [41–43]. Four active sites can exist on the 2×1 surface unit cell of RuO2(110): coordinatively unsaturated Ru (Ruc), bridge O (Obr), full 3-coordinated O (O3f), and coordinatively unsaturated O (Oc). They later calculated the Gibbs free energy for the thermodynamic barrier in the overall CER process (ΔGloss) and revealed that the Oc site had an optimum ΔGloss value of +0.13 eV [43]. They also revealed that upon introducing a PtO2 monolayer on the top of RuO2(110), the reactivity of the Oc site was further improved by lowering the ΔGloss (−0.08 eV).
The optimum thermodynamic adsorption energies of the reaction intermediates over a wide range of catalytic materials can be deduced using Sabatier’s volcano principle. Hansen et al. calculated the adsorption energetics of various sites on rutile (110) precious metal oxides (RuO2, IrO2, and PtO2) for CER and OER (Fig. 3b) [44]. The highest CER activity of RuO2(110) was obtained with the O2cc site (2Oc), where a single *Cl binds to two Oc sites. In contrast, the Oc and Mc sites were found to be the optimum active sites for the CER on IrO2(110) and PtO2(110), respectively.
The CER is typically performed in aqueous electrolytes in the oxidative potential range. Hence, the OER is unavoidably involved as a parasitic competitive reaction during the CER. Fig. 3c shows the most stable surface structure as a function of potential (U), which considers the intermediate of OER, *OH, and that of CER, *Cl [44]. In most metal oxide catalysts, the two intermediates, *Cl and *OH, commonly share the Oc active site, and the adsorption energy on the Oc site can serve as an activity descriptor for both the CER and OER [40–45]. Therefore, the CER and OER are correlated with each other by the scaling relationship, as encountered in other competitive electrochemical reactions such as the four- and two-electron (4e−/2e−) ORR [46,47]. Under these conditions, the optimized active site that binds Cl with the lowest energy can also bind OH with the lowest energy. Unlike 4e−/2e− ORR, the CER/OER scaling relationship involves different adsorbates (*Cl vs. *OH). Hence, the scaling relation can be thermodynamically overcome by generating an active site with relatively larger adsorption energy toward *OH [41,42]. The doping of heteroatoms into precious metal oxide catalysts [8,20] or the introduction of a new class of catalysts have been explored [28,29] to achieve this goal, aspects of which will be described in detail in the next section.
The most prominent example of a doping strategy is the case of DSA [7,8]. Since Beer disclosed DSA in 1967 [48], this catalyst has been the mainstay of CER electrocatalysts for the past half-century [7]. Surprisingly, clear evidence underpinning the improvement in CER selectivity by doping RuO2 with TiO2 is still lacking. Exner et al. theoretically investigated the role of TiO2 coating on RuO2(110) in improving the selectivity of the CER [49]. Fig. 3d presents a volcano plot showing the change in ΔGloss as a function of the Gibbs free energy of the Cl and OH species adsorbed on the Oc site (ΔG0[Oc]). The pure RuO2(110) exhibited a lower ΔG0[Oc] value of 1.43 eV (symbol ×) than TiO2 monolayer coated RuO2(110) (2.88 eV, symbol ○), and the former showed lower ΔGloss values both for the CER and OER than the latter. Interestingly, the ΔGloss of the OER changes more sharply as a function of ΔG0[Oc] than that of the CER. The introduction of the TiO2 monolayer resulted in a larger gap between the ΔGloss values of the CER and OER. This result suggests that the doping method effectively improves the catalytic selectivity for the CER, which is consistent with experimental results [8,20,50]. From a mechanistic viewpoint, breaking the CER–OER scaling relationship via control of the adsorption energy of intermediates is also an effective method [40,45,51]. Once the adsorption energy of OH on the Oc site was weakened, the ΔG for the formation of *OH on the RuO2(110) surface increased markedly, thus enhancing the selectivity for the CER (Fig. 3e) [45].
Exner constructed a CER activity map by combining the conventional volcano principle, overpotential-dependent microkinetics of the rate-determining step (RDS), and electrochemical step symmetry index (Fig. 3f) [52]. Notably, the CER via Cl adsorbate exhibited remarkably high selectivity over a wide pH range, whereas the CER via OcCl adsorbate underwent a decline in selectivity with increasing pH. The currently prevalent CER catalysts (e.g., RuO2 and IrO2) prefer the formation of *OcCl rather than *Cl. Instead, the introduction of metal oxides with weak adsorption energies with oxygen (e.g., PtO2, TiO2, and VO2) can direct the formation of *Cl [52].
2.3 Kinetics analysis approaches
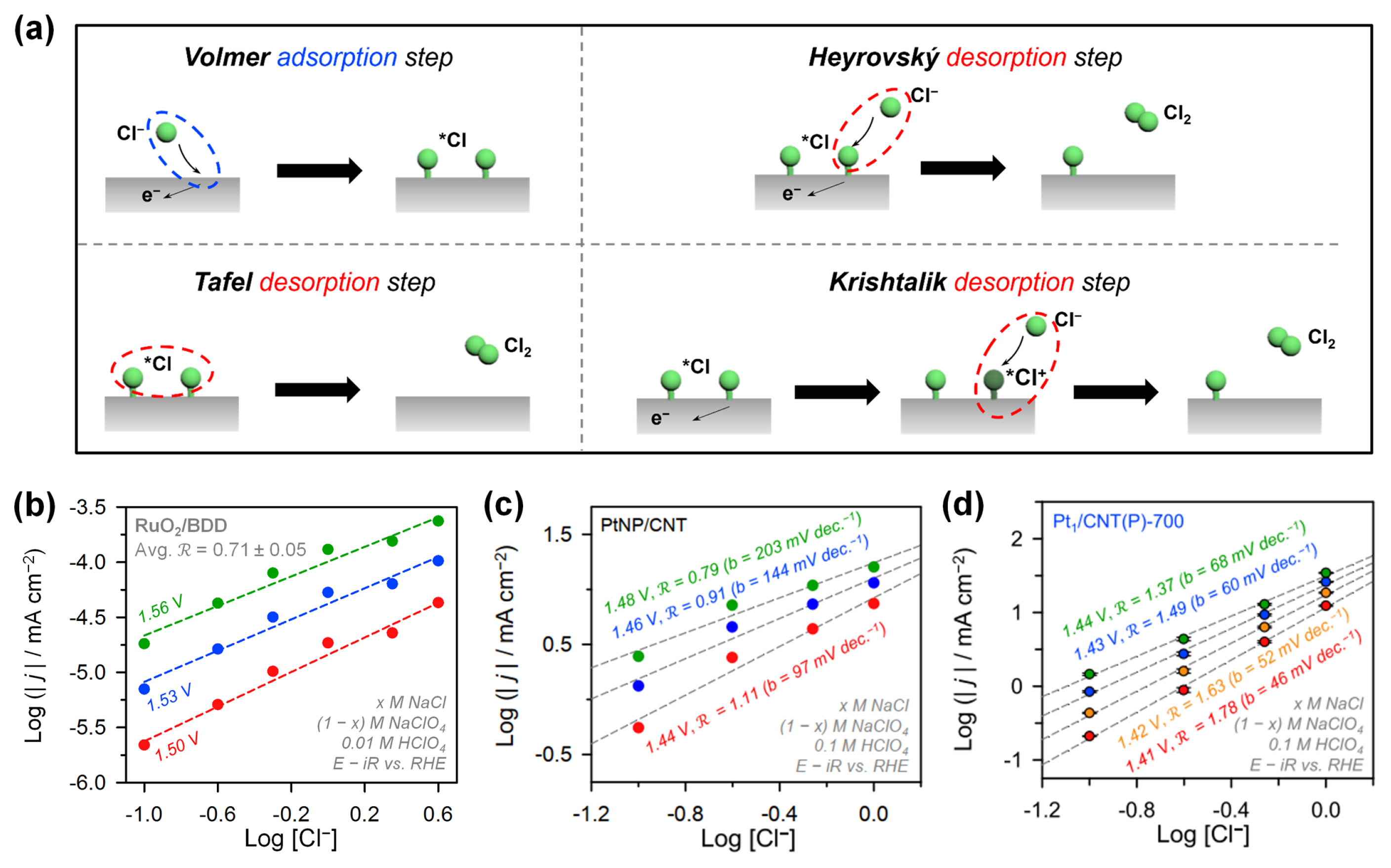
Experimental and theoretical approaches have provided useful guidelines based on Sabatier’s volcano principle for the development of new CER electrocatalysts with enhanced performance. However, the overall electrochemical reaction kinetics are determined by the microkinetics of the elementary reaction steps, which are dependent on the rates of charge transfer [53,54]. The CER involves 2e− charge transfer, similar to the HER, and proceeds via elementary steps of adsorption and desorption [53,55,56]. Accordingly, the microkinetics of the HER have provided an important basis for studying the CER mechanism, and vice versa [53,54,57]. The established CER mechanism comprises one adsorption step and three desorption steps (Fig. 4a). The Volmer step, also known as the Volmer discharge, is the adsorption of Cl− from the electrolyte on the adsorption site (S), forming *Cl (i.e., S–Clads) with one-electron transfer (Equation 5). The three possible desorption steps determine the overall CER mechanism, which can vary depending on the catalyst materials and electrolyte conditions. The most typical desorption step for precious metal oxides is the Heyrovský step in which *Cl is combined with Cl− from the electrolyte to generate a Cl2 molecule (Equation 6). The Heyrovský step resembles Eley–Rideal-type desorption in a heterogeneous gas-phase reaction [58]. The Krishtalik step consists of two sequential reactions: a discharge of *Cl species to form *Cl+ followed by Heyrovský step-like desorption (Equation 7) [55]. The Krishtalik step is only applied for the CER of metal oxides because an electron-rich oxide lattice can stabilize the *Cl+ species. The Tafel step, or Tafel recombination, is a non-electrochemical desorption in which two adjacent *Cl atoms combine to generate a Cl2 molecule without an additional electron (Equation 8). The Tafel step is similar to the desorption step of the Langmuir–Hinshelwood mechanism in heterogeneous gas-phase catalysis [58].
The RDS of metal oxide catalysts can be determined by electrochemical reaction order (ℛ) analyses [53,59], which relates to the number of reactants involved in the RDS. Hence, the reaction order analysis method can serve as a powerful tool for investigating the reaction kinetics of 2e−-transfer reactions, such as CER, as this reaction involves only one reaction intermediate. Ferro and de Battisti prepared RuO2 on the stable and non-reactive boron-doped diamond (BDD) support (RuO2/BDD) and identified the RDS of CER through kinetic analyses (Fig. 4b) [60]. ℛ is defined by the change of current density (j) as a function of the bulk concentration of reactant ([Cl−] for the CER) under constant potential (E) and temperature (T) (Equation 9) [59,61]. The ℛ value is also related to the coverage of the CER intermediate (θCl).
When the CER activity of RuO2/BDD was plotted against [Cl−], its ℛ value in the potential range of 1.5–1.56 V (vs. reversible hydrogen electrode, RHE) was 0.74 ± 0.02. This result suggests that the RDS of the CER over RuO2/BDD is the Heyrovský step involving one reactant (Cl− in the electrolyte), which is consistent with previous results [53,59]. This RDS analysis was carried out by assuming quasi-equilibrium conditions, in which θCl was kept constant (Equation 10). Under such conditions, the current density can be expressed as a function of the reaction rate using the Butler-Volmer equation (Equation 11) and the Ferro-de Battisti test (Equation 12) [60].
where KV, η, F, R, kH, and β are the equilibrium coefficient of the Volmer step, overpotential (E − E0), the Faraday constant, gas constant (8.314 J·K−1·mol−1), rate constant of the Heyrovský step, and the symmetry factor, respectively. Note that β is a charge transfer coefficient for a one-electron transfer step, i.e., an elementary reaction step [62]. For simplicity, a β of 0.5 is generally applied for the medium overpotential range, assuming a symmetric energy barrier [62]. However, the charge transfer of RDS in the CER is likely to be asymmetric, which depicts the large deviation from 0.5, due to its highly complex electrical double layer with evolving Cl2 [62,63]. For example, β = 0.7 was obtained for the CER with the Volmer RDS on Pt–N4 sites [29]. Equation 12 provides a simple test plot based on the experimental values, indicating that exp{(1 − β)·F·η/R·T}·j−1 against exp{−F·η/R·T} should depict a straight slope for the Heyrovský RDS. Recently, Kuo et al. reported the microkinetics of the CER over a RuO2 catalyst in the low current density range (0.001–1 mA cm−2), for which they assumed steady-state conditions instead of a quasi-equilibrium state to reflect very low θCl in this region [64]. Under such conditions, RDS has a ℛ value approaching 2 instead of 1, suggesting that the Volmer step is RDS.
The above analyses indicate that the RDS of the CER should be determined on the basis of a rigorous microkinetics model that can reflect intermediate coverage rather than the slope (b) values of the Tafel equation (Equation 13), which considers simple quasi-equilibrium conditions [53,58–60,65].
Recently, Lim et al. reported platinum-based atomically dispersed catalysts (Pt ADCs) as a new class of CER catalysts. Pt ADCs exhibited different kinetic behaviors from conventional metal oxide catalysts (Fig. 4c,d) [29]. They evaluated the [Cl−]-dependent CER activity of Pt ADCs (Pt1/CNT(P)-700, Fig. 4d) and a Pt-nanoparticle (NP)-based catalyst (PtNP/CNT, Fig. 4c). PtNP/CNT showed an ℛ value of around 1 in the low potential region (1.44–1.48 V vs. RHE), suggesting the Heyrovský step as the RDS. We note that under the CER operating potential, Pt NPs convert to PtOx and behave like RuO2 and IrO2 catalysts. Interestingly, the ℛ of Pt1/CNT(P)-700 approached 2 in a similar potential region with the Volmer RDS (Fig. 4d), suggesting unique reaction kinetics of Pt ADCs that are distinguishable from those of MMO catalysts. These results were further confirmed by in situ X-ray absorption near-edge spectroscopy (XANES) analyses (see Section 3.3).
3. Catalyst Design Strategies for Selective CER
3.1 Foreign metal doped MMO catalysts
The most widely adopted method for improving the CER selectivity of MMO catalysts is doping MMOs with foreign metals. Karlsson and Cornell extensively reviewed the CER selectivity of foreign metal-doped MMO catalysts reported until 2016 [8]. The addition or substitution of Ir [66], Co [67,68], SnO2 [69], Ni [70], Mg [71], and Pt [72] can improve the CER selectivity of MMOs for the OER. In contrast, Fe [73], Zn [74], Zr [75], and Hf [68] were found to decrease the CER selectivity of MMOs. Recent research has focused on developing selective CER catalysts under low [Cl−] and neutral pH conditions, where the CER is thermodynamically unfavorable compared to the OER.
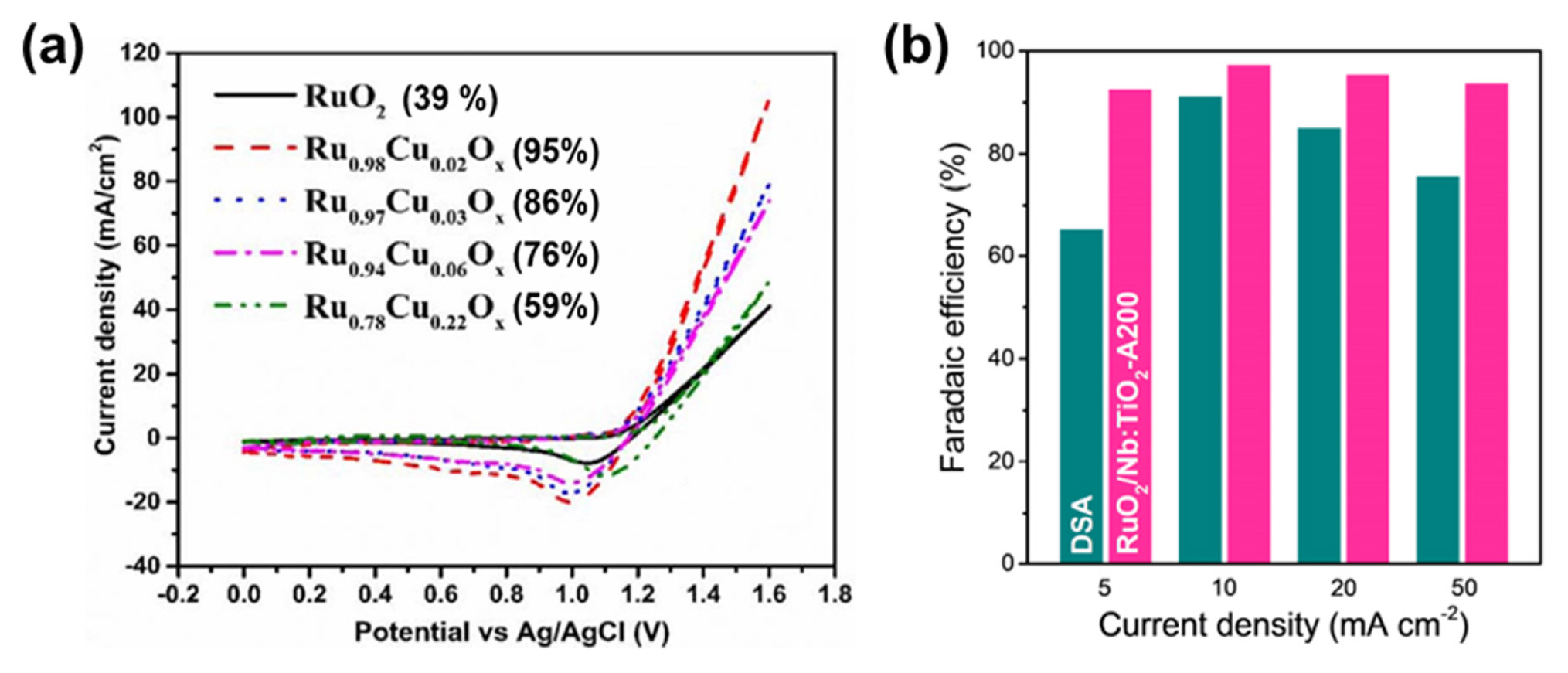
Kishor et al. synthesized Cu-doped RuO2 catalysts by co-electrodeposition of Cu and Ru precursors on a TiO2 electrode and investigated their CER selectivity under neutral pH conditions (0.5 M NaCl, Fig. 5a) [76]. Among the prepared catalysts, Ru99.8Cu0.02Ox exhibited the highest activity (η = 340 mV at 51 mA cm−2), with a high selectivity of 95% (Fig. 5a). The activity and selectivity declined for the catalysts with higher Cu ratios. DFT calculations suggested that the active site of the Ru99.8Cu0.02Ox catalyst is the Obr site in Cu-doped RuO2. This site is energetically unfavorable for the formation of *OH, indicating its high selectivity for the CER.
Lim et al. prepared RuO2 NPs on an Nb-doped TiO2 support (RuO2/Nb:TiO2) using a hydrothermal method, which demonstrated improved CER activity and selectivity via control of the d-band structure of the Ru–Ti interface (Fig. 5b) [77]. In the RuO2/Nb:TiO2 catalyst, the Nb dopant and RuO2 NPs of 20–30 nm were uniformly distributed. The authors suggested that Nb doping allowed the thermal diffusion of Ti atoms into the rutile RuO2 lattice to form the active RuxTi1−xO2 with a thin TiO2 layer, and enhanced the electronic conductivity of TiO2 support. RuO2/Nb:TiO2 exhibited a high CER selectivity of over 90% in a 0.6 M NaCl neutral electrolyte and a high activity with an η of 22 mV at 10 mA cm−2. RuO2/Nb:TiO2 also preserved its initial CER activity after 50 potential cycles. The authors explained that the high CER selectivity of RuxTi1−xO2 could be achieved by decreasing the free adsorption energy of the top O atom via replacing the topmost Ru with Ti. Moreover, the thin TiO2 layer enveloped and protected the active Ru–Ti surface from catalytic degradation. This work also demonstrated that control of the support composition can enhance the durability of the active catalysts.
3.2 Non-Precious Metal Catalysts
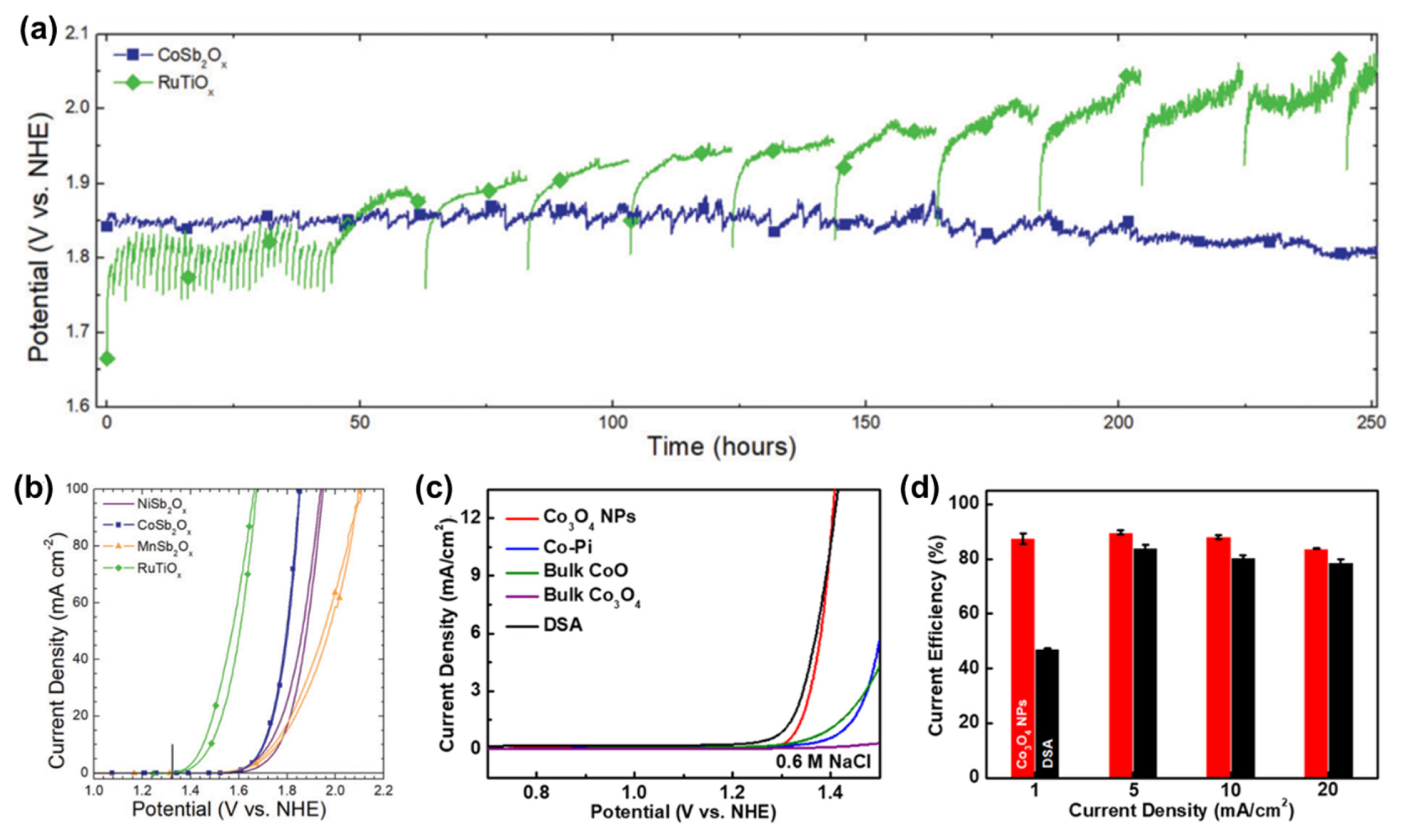
Although DSA and its derivatives exhibit high CER performance, much effort has been devoted to reduce the cost of CER catalysts by developing non-precious-metal-based CER catalysts, such as SnO2–Pb3O4 [24] and VOx–SnOx–SbOx [25]. Moreno-Hernandez et al. prepared transition metal antimonates (MSb2O6, M = Ni, Co, and Mn) with unique crystalline phases via a plasma co-sputtering method [26]. Based on the theoretically predicted Pourbaix diagrams, they confirmed that all MSbOx phases were stable under highly concentrated NaCl and acidic pH conditions. In particular, CoSbOx exhibited very high stability for the CER, preserving a high current density of the CER (100 mA cm−2) for 250 h, which is much superior to the RuTiOx catalyst that showed declining activity during interval tests (Fig. 6a). The catalysts showed initial CER activity in the order RuTiOx > CoSbOx > MnSbOx > NiSbOx (Fig. 6b). Noticeably, CoSbOx exhibited a very high CER selectivity (97.4 ± 3.0%), which was slightly higher than RuTiOx (94.8 ± 0.9%). This work is an excellent example demonstrating that even inexpensive non-precious metal-based catalysts can show high CER activity and selectivity compared to RuO2–TiO2-based catalysts.
Ha et al. exploited Co3O4 NPs as non-precious metal CER catalysts, which showed high CER activity under neutral pH conditions [27]. In the low current density region, Co3O4 exhibited similar CER activity to that of RuO2–TiO2-based DSA (Fig. 6c) and showed a CER selectivity of over 85% (Fig. 6d). However, Co3O4 NPs, in the presence of phosphate (Pi) buffer used to preserve neutral pH, can form Co–Pi species on their surfaces, which are highly active for the OER. Hence, under prolonged operation, the selectivity of Co3O4 NPs rapidly declined [78]. In the in situ XANES study, Co3O4 NPs preserved the oxidation state in the resting state, even under oxidizing CER conditions. This anomalous oxidation state of the Co3O4 NPs suggests that their CER microkinetics are governed by the Krishtalik RDS involving the *Cl+ intermediate. In situ Raman spectra confirmed that H2O was not involved in the reaction under CER conditions. By combining spectroscopic and electrochemical evidence, they suggested Co metallic sites as active sites instead of Oc or Obr sites, which have been suggested as active sites for MMOs.
3.3 Atomically Dispersed Catalysts
ADCs, also termed single-atom catalysts, have recently emerged as a new frontier in heterogeneous catalysis [79–84]. ADCs can take advantage of both homogeneous (~100% utilization of metal sites and high activity and selectivity) and heterogeneous catalysts (recyclability and easy separation). For precious metals, the realization of ADCs can substantially reduce their usage and cost compared to NP-based catalysts while achieving equivalent or superior activity. Importantly, the isolated nature of atomically dispersed sites can promote unique catalytic selectivity by altering the adsorption mode of the reactants compared to NP-based catalysts.
In 2020, Lim et al. reported Pt-based ADC as a highly selective CER catalyst [28] inspired by the high chloride affinity of Pt porphyrin. The Pt ADCs were prepared by physical mixing of a Pt(II) mesotetraphenylporphine (PtTPP) precursor and a carbon nanotube (CNT) support, followed by annealing at 700°C. The resulting Pt1/CNT catalyst consisted of uniformly dispersed, single atomic sites, as clearly visualized by its high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image (Fig. 7a). Extended X-ray adsorption fine structure analysis of Pt1/CNT suggested that the Pt center was coordinated to four nitrogen atoms (Pt–N4 sites), preserving the core structure of the PtTPP precursor after annealing without aggregation. Importantly, Pt1/CNT exhibited superior catalytic activity to RuO2–IrO2–TiO2 DSA and a Pt NP-based catalyst (PtNP/CNT). Pt1/CNT reached a current density of 10 mA cm−2 at an η of 50 mV, which is much lower than those of DSA (η = 105 mV) and PtNP/CNT (η = 120 mV) (Fig. 7b). In the absence of NaCl, Pt1/CNT delivered virtually no current, indicating that Pt1/CNT was inactive for OER. Comparing the CER selectivity of Pt1/CNT and DSA, both catalysts showed similarly high selectivity (> 95%) in an electrolyte of 0.1 M HClO4 and 1.0 M NaCl (Fig. 7c). Interestingly, upon decreasing the concentration of NaCl to 0.1 M, DSA underwent a significant decline in selectivity to around 60%, whereas Pt1/CNT preserved high selectivity. A similar phenomenon was observed for neutral electrolytes. A high selectivity shown by Pt1/CNT suggests that the direct formation of the *Cl intermediate on the Pt–N4 sites can avoid the scaling relation between the CER and OER, which has plagued MMO catalysts.
The origin of the excellent CER activity of Pt1/CNT and the detailed active site structure were investigated by DFT calculations. Three plausible models representing atomically dispersed Pt–N4 sites (PtN4C12, PtN4C10, and PtN2+2C4+4) were constructed (Fig. 7d). Calculation of the adsorption free energies (ΔG’s) of these structures for *Cl (Fig. 7e) indicates that the PtN4C12 site bearing a pyrrolic N moiety is the most plausible active site. This site exhibited the optimum adsorption energy of Cl− but highly unstable adsorption properties with oxygen, corroborating the high CER selectivity of the Pt1/CNT catalyst.
They later demonstrated that catalytically active Pt–N4 sites could also be constructed from a combination of a metal chloride salt (H2PtCl6·6H2O) and an ionic liquid in a bottom-up manner [29]. The resulting Pt1/CNT catalysts also showed similar CER activity and selectivity to the catalyst prepared by the top-down method using the PtTTP precursor, demonstrating the general efficacy of Pt1/CNT catalysts for the CER.
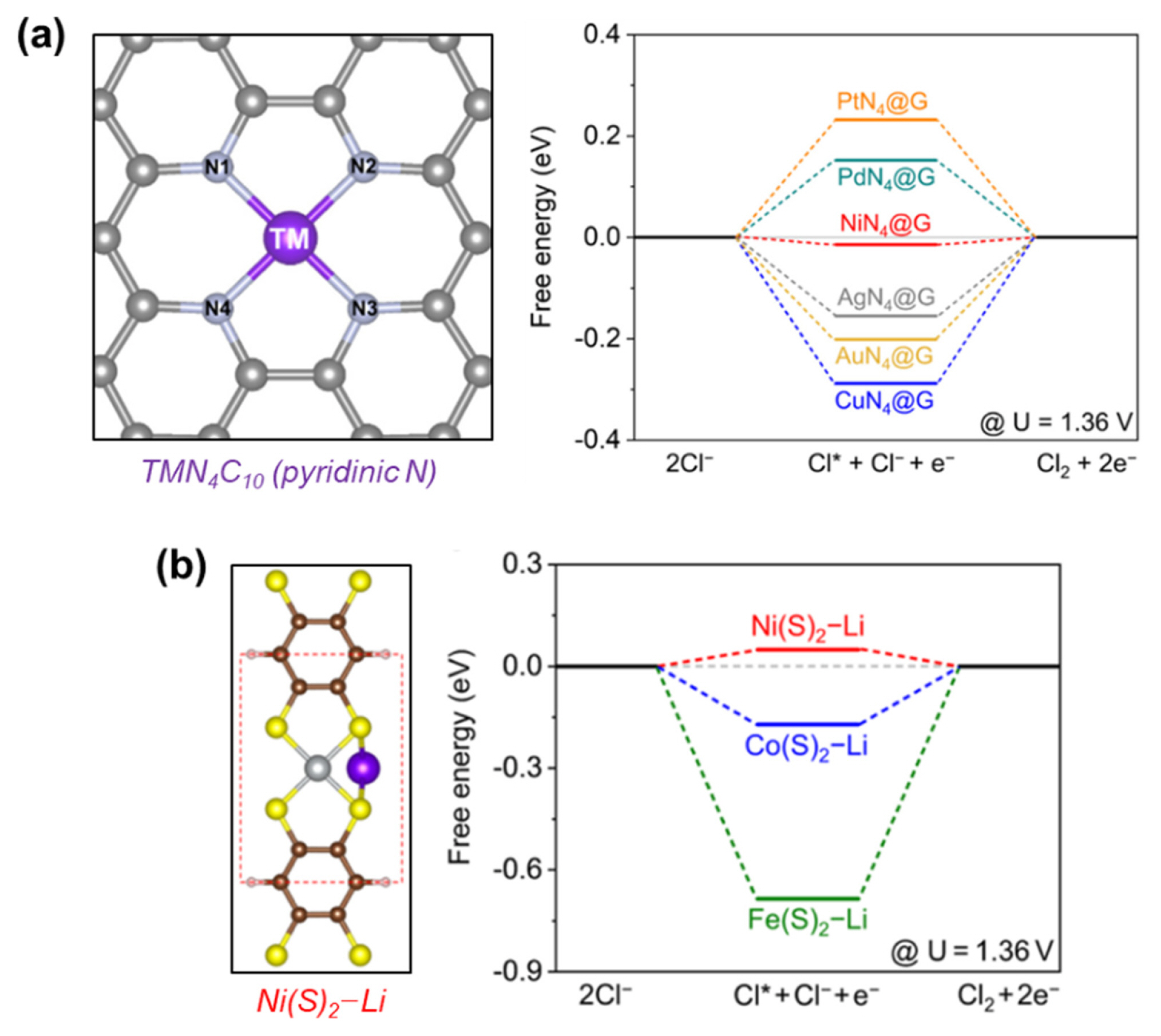
The viability of ADCs as selective CER catalysts was further explored theoretically by DFT calculations. Liu et al. calculated the ΔG’s of *Cl adsorbates on ADCs bearing the MN4C10 structure, which were formed by the coordination between transition metals (Ni, Cu, Pd, Pt, Ag, and Au) and pyridinic N sites (Fig. 8a) [85]. Among the examined structures, NiN4C10 exhibited the lowest ΔG for *Cl formation and was expected to show the highest CER activity. In contrast, NiN4C10 showed a very large overpotential for the formation of *OOH, which is considered the RDS for the OER, suggesting its high selectivity for the CER. The same group also proposed new carbon-based CER catalysts comprising Ni(S)2–Li species as active sites using DFT calculations (Fig. 8b) [86]. Compared to analogous structures containing Co or Fe centers, Ni(S)2–Li exhibited the smallest ΔG for *Cl formation, which originated from a lower p–d hybridization between Cl 2p and Ni 3d than other Fe 3d or Co 3d centers. The resulting weak Ni–Cl interaction of Ni(S)2–Li species could benefit the desorption of Cl2 gas, suggesting its potential utility as a highly active CER catalyst.
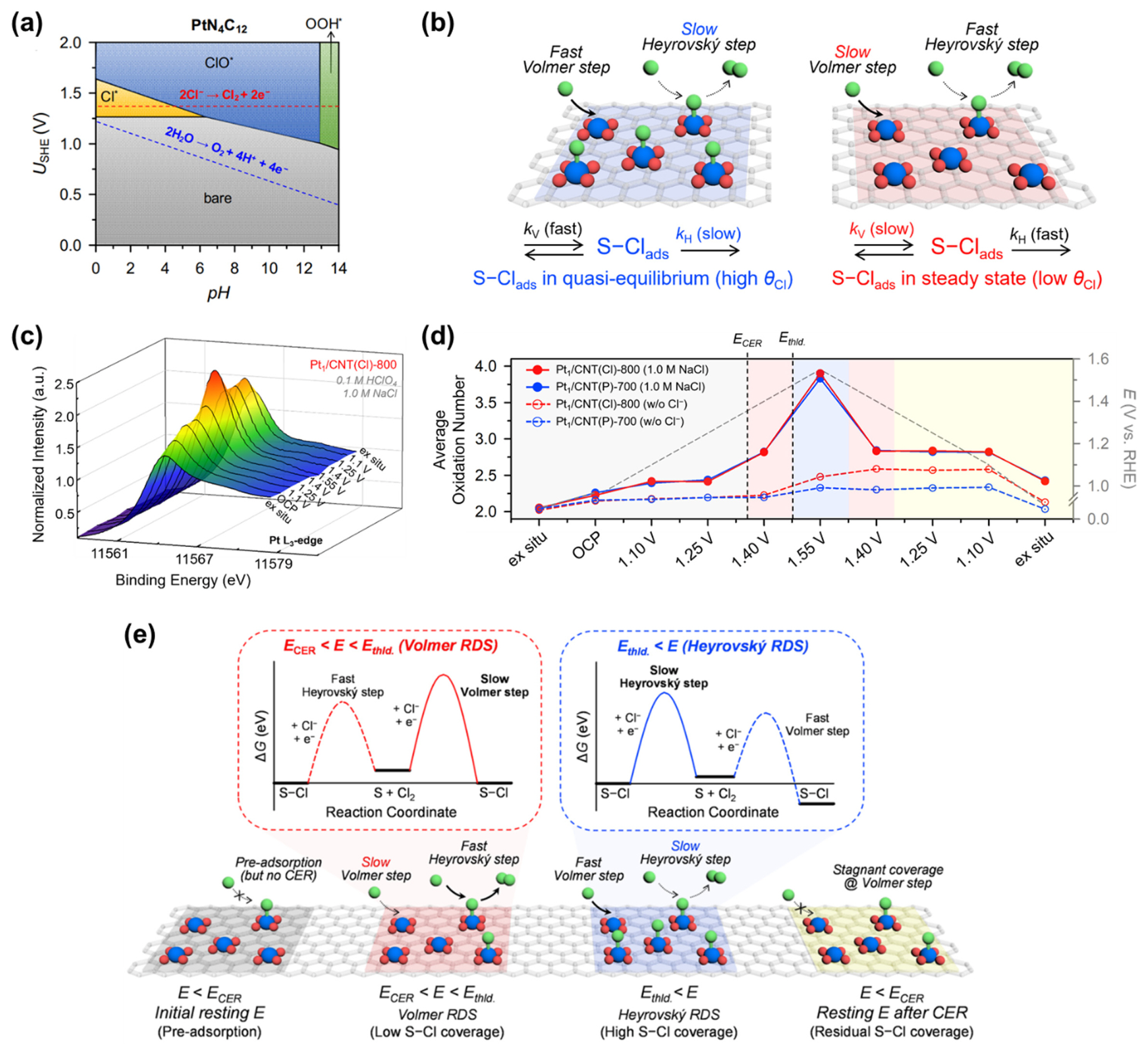
Pt ADCs comprising Pt–N4 active sites are expected to have fundamentally different CER kinetics and mechanisms than MMO catalysts, as the former involves the *Cl intermediate, whereas the *OCl intermediate is prevalent in the latter. Lim et al. combined electrochemical reaction order analysis and in situ XANES analyses to investigate the reaction kinetics and mechanism of Pt1/CNT catalysts in the CER [29]. The PtN4C12 structure, which was identified as the most plausible active site structure for Pt1/CNT [28], exhibits a unique behavior in its Pourbaix diagram (Fig. 9a) [28]; PtN4C12 preadsorbed with *Cl species is more stable than bare PtN4C12, even below the standard reduction potential of CER (1.36 V vs. SHE). Based on previous CER kinetics studies [53–61,64], the CER kinetics of Pt1/CNT catalysts were modeled by considering quasi-equilibrium and steady-state approximations, which are schematically presented in Fig. 9b.
In situ electrochemical XANES spectra can provide valuable information on the redox behavior of the active sites during electrocatalytic reactions. Specifically, the whiteline (WL) intensity of the in situ XANES spectra reveals changes in the relative oxidation state of the active site. The average oxidation number of Pt can be calculated by integrating the area of the WL peak and interpolating the resulting value into those of Pt references with well-known oxidation numbers (e.g., K2PtCl4 for Pt2+ and H2PtCl6 for Pt4+). Importantly, for Pt1/CNT catalysts, the coverage of the reaction intermediate (θCl) on the Pt–N4 active sites can be directly linked to the oxidation state of Pt, as it was demonstrated that no other intermediates are adsorbed on the Pt–N4 sites in the CER-operating potential range [28]. In situ XANES data and potential-dependent oxidation states of the Pt1/CNT catalysts with ascending and descending potentials are presented in Fig. 9c and 9d, respectively. Upon immersing the catalyst in the electrolyte, the average oxidation number of Pt1/CNT was 2.2. With the potential being increased to 1.1 and 1.25 V (vs. RHE), the average oxidation number increased to 2.4, which originates from the preadsorption of Cl− below ECER (Fig. 9a). When the potential was increased to 1.4 and 1.55 V (vs. RHE), the average oxidation number gradually increased to 2.8 and 3.9, respectively. Hence, at a low potential of 1.4 V (vs. RHE), θCl is low, and the reaction proceeds under the steady-state condition, which changes to almost saturated θCl under the quasi-equilibrium state at a higher potential (1.55 V vs. RHE).
By combining of the analyses of reaction order, in situ electrochemical XANES spectra, and theoretical prediction, a schematic free-energy diagram (Fig. 9e, top) and a schematic model showing θCl and RDS during the CER are proposed (Fig. 9e, bottom). Below ECER, the preadsorbed Cl− on Pt–N4 sites makes the Heyrovský step and the Volmer step the first and second steps of the CER, respectively. At the low η region (η = 65–90 mV), the Pt1/CNT exhibited the R value of ~2 and the Tafel slope of ~40 mV dec−1 with low θCl, in which the Volmer step is expected as the RDS. However, as the potential was increased, the R value decreased to ~1 and θCl increased, with the Volmer step gradually becoming faster than the Heyrovský step. Consequently, in the high η region, the CER mechanism switches to the Heyrovský step as the RDS. The switch of the RDS by electrochemical and XANES analyses is also consistent with thermodynamic prediction based on free-energy diagrams [28]. This work provides fundamental insights into the CER catalyzed by atomically dispersed Pt catalysts, which exhibit different reaction kinetics and an unusual potential-dependent switch in the RDS.
4. Summary and Outlook
This review overviewed the fundamental issues in CER and design strategies for selective CER electrocatalysts. Physicochemical, theoretical, and electrochemical methods have consistently identified coordinately unsaturated Oc sites as the active sites of MMO-based catalysts for the CER. Kinetic studies revealed that the CER over most MMOs with an Oc active site follows the Heyrovský RDS. As the Oc site also acts as an active site for the parasitic OER, MMO-based catalysts intrinsically suffer from the scaling relationship between the CER and OER. Hence, the design of advanced CER catalysts has been directed toward improving the activity and selectivity of MMO-based catalysts and overcoming the scaling relationship of MMOs. Representative design strategies have been presented, such as doping of MMOs with foreign metals, development of non-precious metal-based catalysts, and discovery of new types of catalysts, such as ADCs. However, newly developed CER catalysts still have their own limitations. MMO-based catalysts generally have the advantage of longevity, but the parasitic OER is unavoidable due to their oxidic surface nature. Pt–N4 ADCs demonstrated excellent CER activity and selectivity and unique reaction kinetics that were unobservable with MMO-based catalysts. However, at present, Pt–N4 sites are realized only with carbon-based supports, which exhibit vulnerability to electrochemical oxidation resulting in shorter operation time than MMOs. We envisage that further exploitation of unexplored catalytic materials having fundamentally different characteristics from typical oxide alloys, such as intermetallic compounds (or long-range-ordered alloy) [87] and high-entropy alloys [88], have the potential to circumvent the scaling relationship.