1. Introduction
Recently, Na-ion batteries have been considered as a promising alternative to Li-ion batteries because abundance in sodium precursors can lead to a cost reduction [1ŌĆō4]. In addition, the existence of a database of electrode materials for Li-ion batteries can promote the development of electrode materials for Na-ion batteries, since the chemistry of Na-ion batteries is similar to that of Li-ion batteries. Drawing inspiration from previously reported cathode materials for Li-ion batteries, many research groups have reported various cathode materials for Na-ion batteries. These include NaxCoO2 [5,6], NaxCrO2 and NaxNiO2 [7], ╬▒-NaFeO2 [8], NaxVO2 [9, 10], Na0.44MnO2 [11], Na2/3[Fe1/2Mn1/2]O2 [12], Na1-xNi0.5Mn0.5O2 [13], NaCo1/3Ni1/3Mn1/3O2 [14], NaMPO4 (M=Fe, Mn) [15], A2MPO4F (A = Na, Li, M = Fe, Mn, Co, Ni) [16], NaVPO4F [17], Na1.5VOPO4F0.5 [18], Na3V2(PO4)2F3 [19,20], Na3Fe3(PO4)4 [21], Na2FeP2O7 [22], t-Na2(VO)P2O7 [23], and Na2MnP2O7 [24,25]. Our group also reported a new pyrophosphate cathode material (triclinic Na4-╬▒M2+╬▒/2(P2O7)2 where 2/3 Ōēż ╬▒ Ōēż 7/8 and M = Fe, Fe0.5Mn0.5, or Mn) and showed promising electrochemical performance including negligible capacity fading over 60 cycles and good rate performance [26]. Then, Niu et al. and Chen et al. optimized the electrochemical performance of Na3.12Fe2.44(P2O7)2 [27,28]. However, its specific energy (3.0 V ├Ś ca. 85 mA h gŌłÆ1) is still smaller than that of LiFePO4 (3.4 V ├Ś ca. 160 mA h gŌłÆ1), which is used in Li-ion batteries. Not only Na3.12Fe2.44(P2O7)2 but the majority of cathode materials for Na-ion batteries display lower energy densities when compared to those used in Li-ion batteries; thus, an improvement in energy density is vital for commercialization of Na-ion batteries. This can be achieved by either increasing the reversible capacity or the redox potential. It is well known that the redox potential of Co2+/Co3+ in phosphate-based materials for Li-ion batteries is higher than those of Fe2+/Fe3+ and Mn2+/Mn3+ [29]. Therefore, in this report, we introduce the Co analogue (Na3.12Co2.44(P2O7)2) of triclinic Na3.12Fe2.44(P2O7)2 as a high-voltage cathode material for the improvement in energy density. Moreover, triclinic Na3.12Co2.44(P2O7)2 was also compared with orthorhombic Na2CoP2O7 to investigate the effect of crystal structure on electrochemical performance. It was known that three crystal structures of sodium cobalt pyrophosphates (Na2CoP2O7) exist: orthorhombic (Pna21), tetragonal (P42/mmm), and triclinic (PŌłÆ1) structures [30ŌĆō32]. Whereas the electrochemical performance of nanosized orthorhombic Na2CoP2O7 has been reported [33], the synthesis and electrochemical performance of triclinic Na3.12Co2.44(P2O7)2 have not yet been studied to the best of our knowledge. Furthermore, we demonstrate that Na3.12Co2.44(P2O7)2 has an appropriate channel size for Na+ ions through comparison of the electrochemical behaviors of Na3.12Co2.44(P2O7)2 in Na-ion and Li-ion cells.
2. Experimental
2.1 Synthesis
Triclinic Na3.12Co2.44(P2O7)2 and orthorhombic Na2CoP2O7 were synthesized through a conventional solid-state method. Stoichiometric amounts of sodium carbonate (Na2CO3, Sigma-Aldrich, Ōēź 99.5%), cobalt nitrate hexahydrate (Co(NO3)2┬Ę6H2O, Sigma-Aldrich, 98+%), and ammonium phosphate dibasic ((NH4)2HPO4, Sigma-Aldrich, 98+%) were mixed, and then heated at 300┬░C for 6 hours and 600┬░C for 12 hours in Ar atmosphere.
2.2. Chemical de/sodiation processes
Chemical oxidations of Na3.12Co2.44(P2O7)2 (as well as the electrochemical charging processes) to Na3.12-x Co2.44(P2O7)2 (x = ~1) and Na3.12-yCo2.44(P2O7)2 (y = ~2) is performed using nitronium tetrafluoroborate (NO2BF4, 95+%, Sigma-Aldrich) with equimolar amounts, stirred with acetonitrile (AN) solvent for 3 hours, then filtered and washed with ethanol. In the same way, sodium iodide (NaI, Ōēź 99.5%, Sigma-Aldrich) is added for chemical reduction (electrochemical discharging processes) with AN, stirred for 5 hours. Changes in atomic composition of Na3.12Co2.44(P2O7)2 were estimated using an inductively coupled plasma-mass spectrometer (ICP-MS, Perkin-Elmer NexION 350D).
2.3 Materials characterization
Powder X-Ray diffraction (XRD) data were collected on a Bruker D2 Phaser diffractometer using Cu-K╬▒ radiation (╬╗=1.5418 A) operated from 2╬Ė = 10 ŌĆō 80┬░. Rietveld refinements were performed using powder XRD patterns with the TOPAS program. The morphologies of powders were examined using the Hitachi S-4800 Field Emission Scanning Electron Microscopy (FE-SEM).
2.4 Electrochemical characterization
Each active material (70 wt.%) was mixed with carbon black (Super-P, 15 wt.%) and polyvinylidene fluoride (PVdF, 15 wt.%) in 1-methyl-2-pyrrolidinone solvents (NMP, Sigma-Aldrich, anhydrous, 99%). The slurries were coated on Al foil as a current collector. Electrochemical performances of Na3.12Co2.44(P2O7)2 and Na2CoP2O7 were examined using coin cells (CR2032 type). Na and Li metals were used as counter electrodes for Na-ion and Li-ion half-cells, respectively. A 0.8 M solution of sodium hexafluorophosphate (NaPF6) dissolved in a mixture (1:1, v/v) of ethylene carbonate (EC) and propylene carbonate (PC), with 5 wt.% of fluoroethylene carbonate (FEC) and 1 wt.% of tris(trimethylsilyl)phosphite (TMSP) additives, and 1.3 M of lithium hexafluorophosphate (LiPF6) dissolved in a mixture (3:2:5, v/v/v) of ethylene carbonate, ethyl methyl carbonate (EMC), and diethyl carbonate (DEC) were used as electrolytes for Na-ion and Li-ion half-cells, respectively. Galvanostatic experiments were performed at the current density of 4.75 mA gŌłÆ1 (ca. 0.05 C-rate) in the voltage ranges from 2.4 to 4.5 V vs. Na/Na+ and from 2.7 to 4.8 V vs. Li/Li+. Furthermore, the potentiostatic intermittent titration technique (PITT) was performed to estimate diffusion coefficients. The potential step of 10 mV was applied at various depth of discharge states. Each potential step was applied until the current density was less than 0.75 mA gŌłÆ1. Electrochemical impedance spectroscopy (EIS) was carried out in the frequency range from 300 kHz to 1 mHz with voltage amplitude of 5 mV using the BioLogic SP-150.
3. Results and Discussion
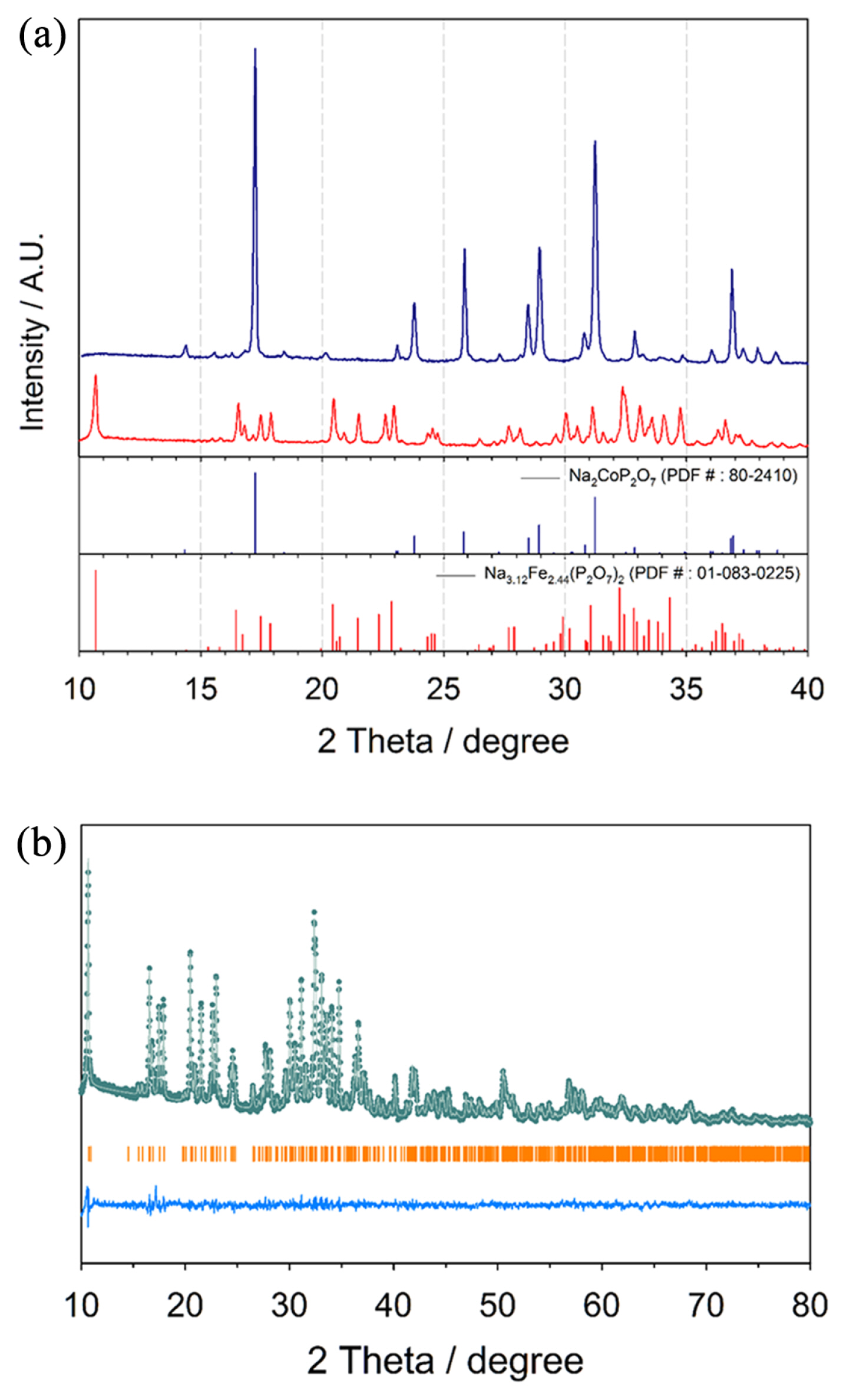
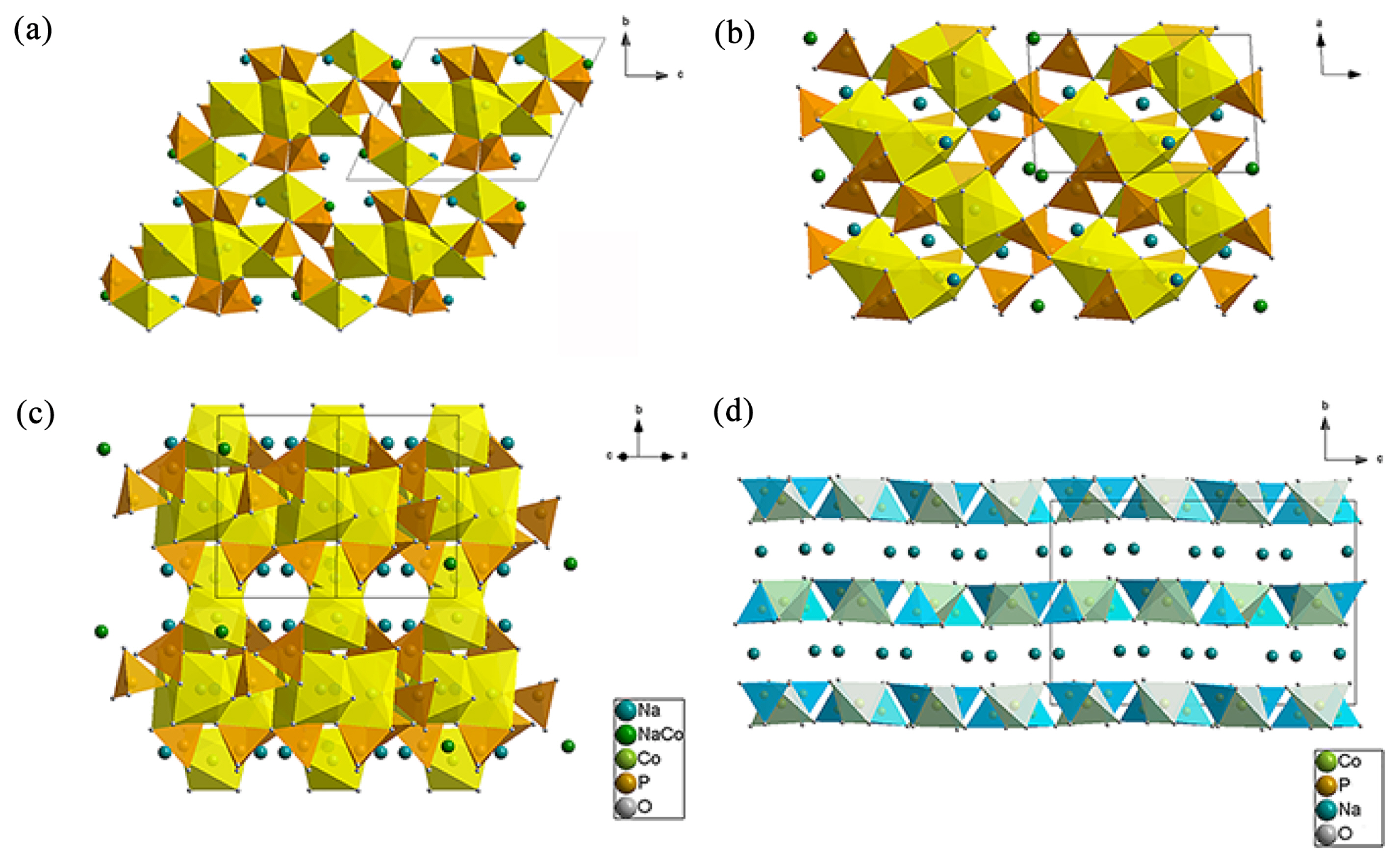
Two types of sodium cobalt pyrophosphates, triclinic Na3.12Co2.44(P2O7)2 and orthorhombic Na2CoP2O7, were synthesized through a conventional solid-state method via heating at 600┬░C under Ar. Fig. 1(a) shows the X-ray diffraction (XRD) patterns of Na3.12Co2.44(P2O7)2 and Na2CoP2O7, indicating that they contained no impurities. We performed the Rietveld refinement to determine the lattice parameters of Na3.12Co2.44(P2O7)2 (Fig. 1(b) and Table 1). This revealed that Na3.12Co2.44(P2O7)2 has a crystal structure similar to triclinic Na3.12Fe2.44(P2O7)2. However, a slight distortion was observed due to the different ionic size between Co and Fe. As shown in the crystal structure images (Fig. 2(a), (b), and (c)), Na3.12Co2.44(P2O7)2 is composed of two centrosymmetrical crowns of Co2P4O20 and Co2P4O22, and has three dimensional diffusion channels along the [100], [010], and [101] directions. On the other hand, orthorhombic Na2CoP2O7 has a layered crystal structure having a two-dimensional diffusion path perpendicular to the [010] direction (Fig. 2(d)).
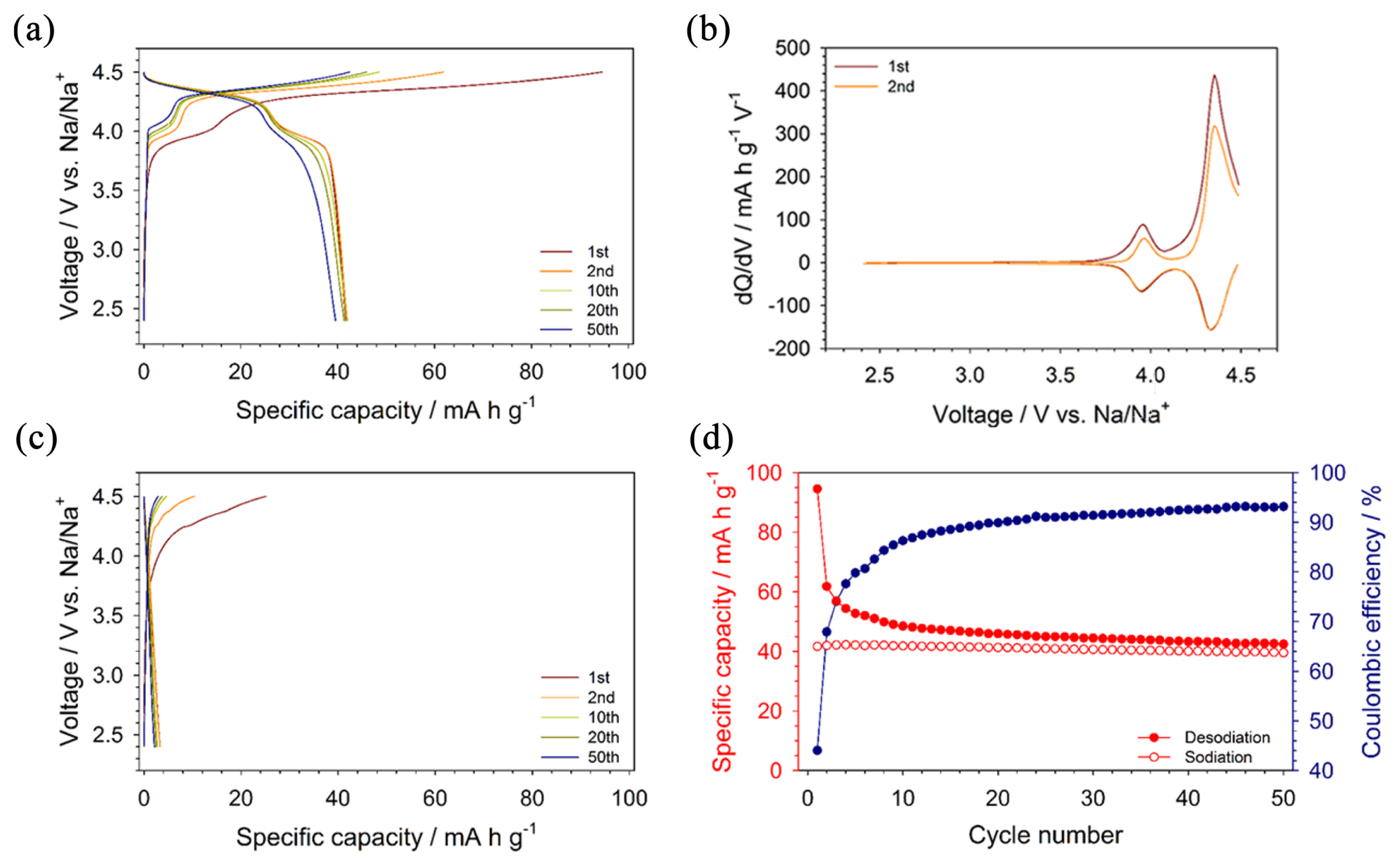
We compared the electrochemical performances of triclinic Na3.12Co2.44(P2O7)2 and orthorhombic Na2CoP2O7, as shown in Fig. 3. Na metal and a 0.8 M solution of NaPF6 dissolved in a mixture solvent of EC and PC 1:1 (v/v) were used as a counter electrode and electrolyte, respectively. FEC and TMSP were added as electrolyte additives to improve the electrochemical stability of electrolytes at high voltages [34]. As shown in the voltage profiles and dQ/dV plots of Na3.12Co2.44(P2O7)2 (Fig. 3(a) and (b)), two plateaus were observed at very high redox potentials of ~3.9 and 4.3 V vs. Na/Na+. This implies that Na3.12Co2.44(P2O7)2 exhibited a higher redox potential than Na3.12Fe2.44(P2O7)2 (~3.0V vs. Na/Na+). Although Na3.12Co2.44(P2O7)2 delivered a smaller reversible capacity (approximately 40 mA h gŌłÆ1) than Na3.12Fe2.44(P2O7)2 (~ 85 mA h gŌłÆ1), this is attributed to the upper limit of the charging voltage (4.5 V vs. Na/Na+) due to the severe electrolyte decomposition above 4.5 V. Na3.12Co2.44(P2O7)2 also showed excellent reversibility with negligible capacity fading over 50 cycles, although the coulombic efficiency was not sufficiently high because of electrolyte decomposition (Fig. 3(d)). In contrast to triclinic Na3.12Co2.44(P2O7)2, layered Na2CoP2O7 did not deliver any observable reversible capacity in the voltage range between 2.4 and 4.5 V vs. Na/Na+ (Fig. 3(c)). The poor electrochemical activity of Na2CoP2O7 is attributed to very slow kinetics resulting from the high activation energy for the diffusion of Na+ ions due to the small channel size (~3.1 A); note that the channel size of Na3.12Co2.44(P2O7)2 is ~4.2 A. Since Na3.12Co2.44(P2O7)2 and Na2CoP2O7 have similar particle sizes that ranges from hundreds of nanometers to a few micrometers, as shown in the SEM images (Fig. 4), the different electrochemical activities of Na3.12Co2.44(P2O7)2 and Na2CoP2O7 were not due to the difference in the particle sizes.
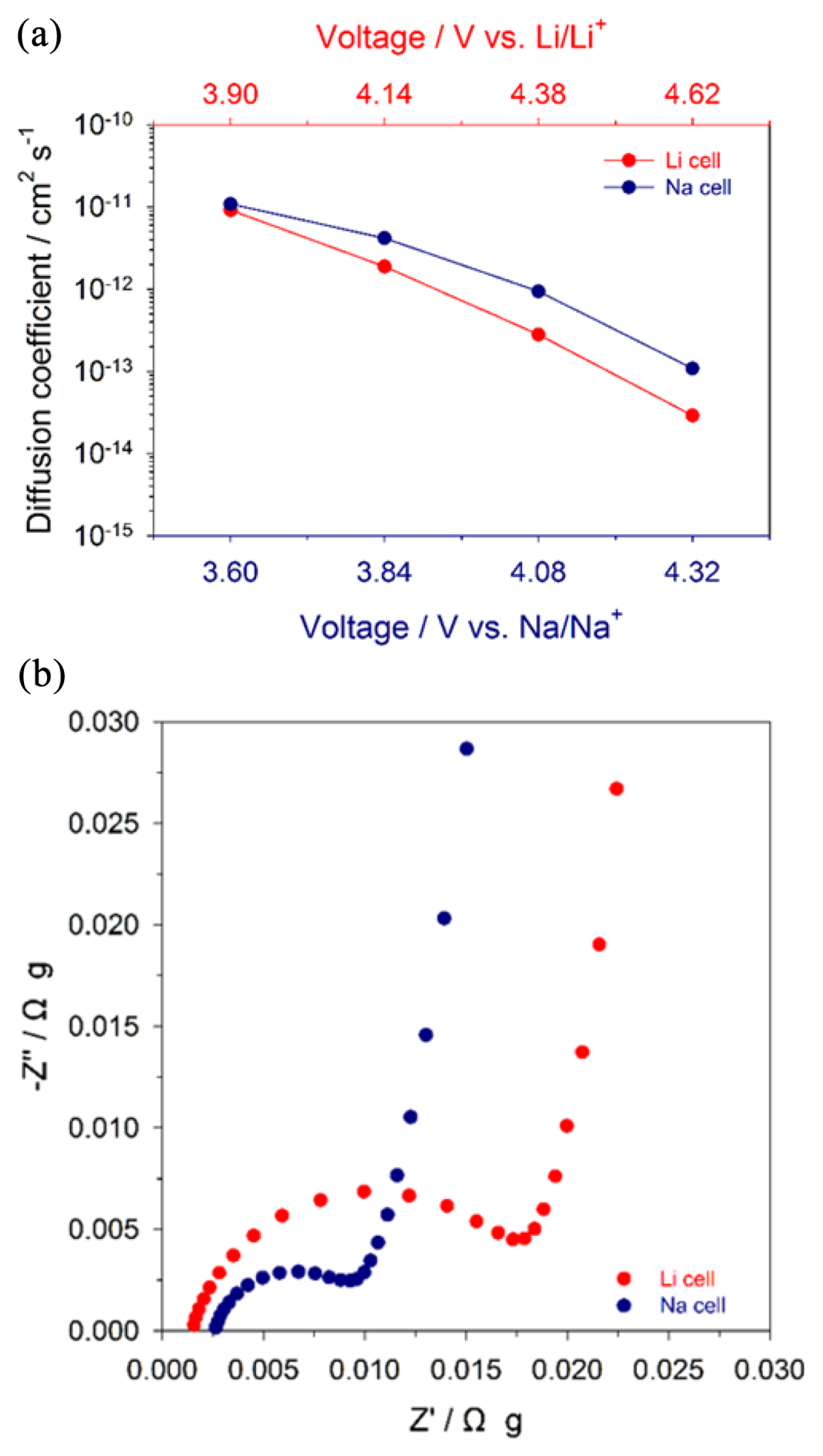
Moreover, Na3.12Co2.44(P2O7)2 and Na2CoP2O7 were examined as cathode materials for Li-ion batteries (Fig. 5). Li metal and a 1.3 M solution of LiPF6 dissolved in a mixture solution of EC: EMC: DEC (3:2:5, v/v/v) were used as a counter electrode and electrolyte, respectively. Li-ion cells of Na2CoP2O7 still exhibited no electrochemical activity in the voltage range between 2.7 and 4.8 V vs. Li/Li+ (Fig. 5(c)), which is consistent with the result of Na-ion cells. Li-ion cells of Na3.12Co2.44(P2O7)2 delivered a reversible capacity of approximately 40 mA h gŌłÆ1 (Fig. 5(a)), which is similar to its Na-ion cells. However, when compared to the performance for Na-ion cells, Na3.12Co2.44(P2O7)2 exhibited a larger polarization and poorer capacity retention for Li-ion cells, as shown in the dQ/dV plots (Fig. 3(b) and 5(b)). It is notable that the overpotential of Na-ion cells was smaller than that of Li-ion cells, despite the fact that Na+ ions are larger than Li+ ions. To understand this behavior, diffusion coefficients and charge transfer resistances were investigated using PITT and EIS, respectively (Fig. 6). The diffusion coefficients (D) were calculated using the equation below with the assumption of the finite diffusion to exclude the effect of surface roughness of particles:
where n is an equivalent amount of electrons, F is Faraday constant, A is the surface area of particles, ╬öC is the concentration gradient, and a is a particle radius. The diffusion coefficients of Li+ and Na+ ions in Na3.12Co2.44(P2O7)2 were measured using current transients obtained from the PITT (Fig. 6(a)) [35ŌĆō37]. Diffusion coefficients were calculated from the slopes (ŌłÆŽĆ2D/a2) of ln(imp) vs. t. Interestingly, the diffusion coefficients of Na+ ions were higher than those of Li+ ions in Na3.12Co2.44(P2O7)2. This suggests that the large channel size of Na3.12Co2.44(P2O7)2 is more suitable for larger Na+ ions than Li+ ions. This behavior is consistent with Na+-conducting ╬▓-alumina [38ŌĆō40]; the activation energy of Na+ ions for ionic diffusion in ╬▓-alumina is lower than that of Li+ ions. This is attributed to the fact that the smaller Li+ is less stable in large sites with a high coordination number, resulting in stronger binding with oxygen than that in the case of Na+. In addition, to compare the charge transfer resistances of Na3.12Co2.44(P2O7)2 between Na cells and Li cells, we performed the EIS analysis using symmetric cells comprising two identical fully charged electrodes. This revealed that Naion cells have a smaller charge transfer resistance than Li-ion cells (Fig. 6(b)). For these reasons, Na3.12Co2.44(P2O7)2 showed better electrochemical performance for Na-ion batteries, indicating that the structure of Na3.12Co2.44(P2O7)2 is more suitable for Na-ion batteries.
Since the high charging voltage inevitably leads to the oxidative decomposition of the electrolyte, the electrochemical reaction mechanism of Na3.12Co2.44(P2O7)2 was investigated through chemical sodiation and desodiation. The redox potentials of oxidizing and reducing agents must be above and below a redox potential of Na3.12Co2.44(P2O7)2 (ca. 3.9 and 4.3 V vs. Na/Na+), respectively. For this reation, NO2BF4 (V (NO2+/NO2) = ca. 4.8 V vs. Na/Na+) and NaI (V (I2/IŌłÆ) = ca. 3.1 V vs. Na/Na+) were used for chemical desodiation and sodiation, respectively [41]. The sequential formation of Na3.12Co2.44(P2O7)2 ŌåÆ Na2.5Co2.44(P2O7)2 ŌåÆ Na2.0Co2.44(P2O7)2 ŌåÆ Na2.65Co2.44(P2O7)2 ŌåÆ Na2.75Co2.44(P2O7)2 was obtained via repetitive desodiation and sodiation using equimolar NO2BF4 and NaI, respectively. The amount of Na and Co in Na3.12-xCo2.44(P2O7)2 was measured by ICP analysis (Fig. 7(a)). Then, we obtained the XRD patterns of those compositions, as shown in Fig. 7(b). The XRD peaks gradually and reversibly shift as x in Na3.12- xCo2.44(P2O7)2 increased and decreased, and we also observed no evidence for a two-phase mixture of the end member phases. This implies that the Na3.12Co2.44(P2O7)2 electrode proceeds in a one-phase reaction. This solid solution behavior of Na3.12Co2.44(P2O7)2 was also confirmed by the appearance of sloping voltage profiles (Fig. 3(a)).
4. Conclusions
We synthesized two types of sodium cobalt pyrophosphates, triclinic Na3.12Co2.44(P2O7)2 and orthorhombic Na2CoP2O7, through a conventional solid-state method via heating at 600┬░C under Ar. Na3.12Co2.44(P2O7)2 and Na2CoP2O7 were examined as high-voltage cathode materials for Na-ion batteries. Whereas Na2CoP2O7 showed no electrochemical activity, Na3.12Co2.44(P2O7)2 exhibited good electrochemical performance, such as high redox potential at ca. 4.3 V (vs. Na/Na+) and stable capacity retention over 50 cycles, although Na3.12Co2.44(P2O7)2 delivered approximately 40 mA h gŌłÆ1. This is attributed to the fact that Na2CoP2O7 (~3.1 A) has smaller diffusion channel size than Na3.12Co2.44(P2O7)2 (~4.2 A). Moreover, we compared the electrochemical performances of Na3.12Co2.44(P2O7)2 between Na cells and Li cells. Na cells showed smaller overpotential than Li cells. This is due to the fact that Na3.12Co2.44(P2O7)2 displayed a smaller charge transfer resistance and higher diffusivity for Na+ ions when compared to Li+ ions. This implies that the large channel size of Na3.12Co2.44(P2O7)2 were more appropriate for Na+ ions than Li+ ions. Therefore, Na3.12Co2.44(P2O7)2 is considered a promising candidate for a high-voltage cathode material. We believe that the high energy density of Na-ion batteries can be achieved with Na3.12Co2.44(P2O7)2 if stable electrolytes above 4.5 V vs. Na/Na+ are developed.