1. Introduction
Aprotic lithium-oxygen batteries are promising next-generation electric power sources due to their eco-friendly and sustainable nature as well as high theoretical capacity of 5,200Wh/kg, which is almost 5-10 times larger than state-of-the-art lithium ion battery system [1-5]. Remarkably, such high energy densities are attributed to their discharge reactions between the lithium and oxygen fuel which produces Li2O2 ideally [6,7]. Despite of the exceptional capacities, these lithium-oxygen batteries have several challenges such as a low round-trip efficiency [8,9] and poor cycleability [10,11]. These problems are quite severe and may result from the thermodynamically stable discharge products and its insulating character, which means that much higher energy should be exhausted during charging to dissociate them, and the polarization cannot but be severely increased during the whole ORR(Oxygen Reduction Reaction)/ OER(Oxygen Evolution Reaction). In an attempt to overcome such a significant prevailing drawbacks, much efforts have been paid towards designing new electrode architecture and finding a promising material that acts as an efficient catalyst enabling a stable cyclic retention [12-15]. When the ORR and OER are considered during discharge/charge, the main issue can fall into a large volume contact between catalyst and discharge product enabling a reversible dissociation of discharge product and maintaining the efficient electrical connection with catalysts. Recently, Bruce et al. suggested that the choice of catalyst materials is crucial and, MnOx polymorphs can evolve various capacities and cyclic stabilities due to their special polymorphic crystal structures and higher surface areas [16]. Therefore, the nano sized materials which have been shown to possess much high surface area, have been used for the processing of efficient catalyst eg. Fe2O3, MnO2, Co3O4, etc. since they can give large surface-to-volume ratio, may shortens Li ion and O2 gas diffusion length, and easily ensure e- contact point between catalysts and discharge product to facilitate the electrochemical reaction of electrode [17-20]. However, using a TiO2 nanoparticles for the catalyst of lithium-oxygen batteries has been rarely reported in the literature even though it has promising attributes and have been widely investigated for the catalysts of various devices owing to its low cost, non-toxicity, eco-friendliness, and natural abundance [21-25].
In this regard, we here report on TiO2 nanoparticles (NPs) as a cathode catalyst material, which is expected to provide enough contact area to maximize the electrochemical properties of lithium-oxygen cell such as round-trip efficiency, rate capability and cyclic retention.
Experimental
The synthesis of TiO2 NPs was carried out by using a hydrothermal method. The titanium butoxide (97%, Sigma-Aldrich) and Benzyl alcohol (99%, Sigma-Aldrich) were used as received without any further purification. In a typical hydrothermal synthesis, firstly titanium butoxide (5 g) and benzyl alcohol (20 mL) were placed in a 50 mL glass bottle and mixed by vigorous stirring at room temperature for 1 h in ambient. The mixed solution were then transferred to a 100 mL Teflon-lined stainless-steel autoclave and heated to 220℃ for 12 h. After naturally cool down to room temperature, the obtained precipitates were collected by centrifugation and washed several times with the deionized water and ethanol. Then the samples were dried at 60℃ for 12 h in a vacuum oven.
The electrodes in this study were fabricated by a mixing TiO2 catalyst nanoparticles, Ketjen black (EC600JD), and PVDF-HFP copolymer with a weight ratio of 40:45:15 by using N-methylpyrrolidone (NMP) as a solvent. The resulting slurries were pasted onto the carbon paper (GDL; Gas diffusion layer) and then dried in a vacuum oven at 120℃ for 5 h. After drying, the electrochemical properties of the prepared electrodes were evaluated by using Swagelok-type cells assembled in an argon-filled glove box. Lithium metal foils were used as a counter electrode, glass fiber disk as the separator and 1M LiCF3SO3 solution in tetraethyleneglycol dimethylether (TEGDME) as the electrolyte. Subsequently, the sealed cell was removed from the argon-filled glove box and then purged with O2 gas (99.995%) for 10 min at 1 sccm, galvanostatically charged and discharged at a current density of 200 mA·g–1carbon over a range of 2.0 - 4.5 V.
The prepared TiO2 NPs were characterized by an XRD (Bruker/New D8 Advance, diffractometer using a graphite-mono-chromatized Cu Kα radiation at 40 kV and 40 mA) and transmission electron microscope (TEM; Omega EM912, operated at 120 kV).
Results and Discussion
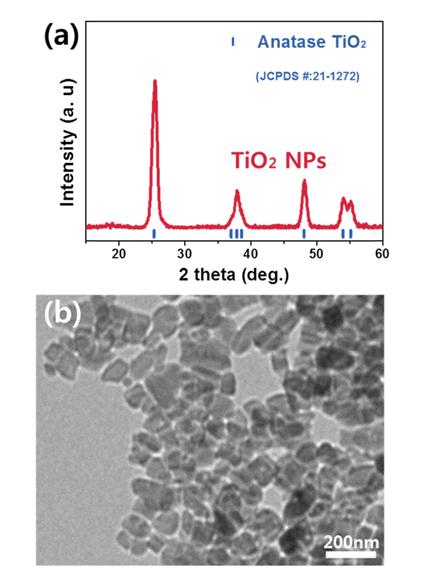
Fig. 1 shows the crystallinity and morphology of hydrothermally synthesized TiO2 NPs through a X-ray diffraction (XRD) pattern and a transmission electron microscopy (TEM) image. The X-ray diffraction pattern in Fig. 1a indicates that the TiO2 NPs were well crystallized into tetragonal structure with dominant diffraction peaks at 2 theta of 25.2°, 38.1°, 48.1° and 54.1°/55.2° corresponding to (101), (004), (200) and (105)/(211) and engaged in the space group I41/amd [26]. Moreover, there were not any obvious impurity or rutile/brookite peaks and peak shift related to lattice change. The representative TEM image in Fig. 1b clearly shows that the TiO2 NPs were in rectangular shape with a diameter of about 100 nm, which can provide large surface area for more facile chemisorption and better accommodation of the discharge products.
Fig. 1.
(a) X-ray diffraction pattern of hydrothemally synthesized TiO2 nanoparticles and (b) their transmission electron microscopic image.
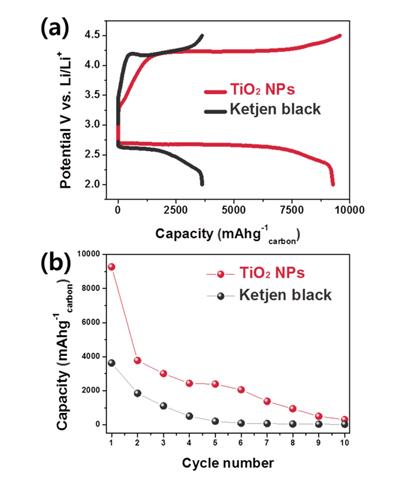
Electrochemical performances of the lithium-oxygen batteries including TiO2 NPs as a cathode catalyst are shown in Fig. 2. The charge-discharge measurements were carried out in the voltage range of 2.0 - 4.5 V at room temperature. The initial galvanostatic discharge-charge profiles are presented in Fig. 2a, which demonstrates that the TiO2 NPs have obvious superior catalytic activity for the formation of lithium oxides such as Li2O2, Li2O and its dissociation as indicated by the attained high capacity and relatively low overpotentials compared to the catalystfree cell. The discharge capacity of lithium-oxygen cell with TiO2 NPs was about 9,200 mAh/g, which is almost 3 times higher than that of catalyst-free lithium-oxygen cell (3,800 mAh/g). In addition, its overpotential was also significantly lower than that of the catalyst-free cell. When the cycleability of lithium-oxygen cell with TiO2 NPs were compared with that of the catalyst-free cell between 2.0 and 4.5 V, lithium-oxygen cell with TiO2 NPs retained relatively high reversible specific capacities over 2,500 mAh·g–1carbon up to the sixth cycles, whereas the catalyst-free cell underwent a drastic capacity decay as shown in Fig. 2b. This may be due to the influence of better catalytic activity of TiO2 and good discharge capability products for lithium-oxygen batteries.
Fig. 2.
The electrochemical performances of lithium-oxygen cells utilizing TiO2 NPs as cathode catalyst and their comparison with the catalyst-free lithium-oxygen cells; (a) The initial discharge-charge curves, (b) The cyclic retention obtained between 2.0 and 4.5 V at a current density of 200 mA·g–1carbon.
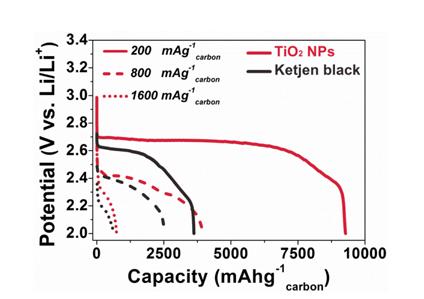
In order to confirm and visualize the detailed effect of TiO2 NPs catalyst on the reaction kinetics of Li-air cells, the rate capability of cathode containing TiO2 NPs and catalyst-free cathode have been tested and compared at higher discharge current densities of 200 mA·g–1carbon, 800 mA·g–1carbon, 1,600 mA·g–1carbon between 2.0 V and 3.2 V, respectively. Lithium-oxygen cell with TiO2 NPs still possess a high discharge capacity of 4,000 mAh·g–1carbon, which is 4 times higher than the current density of initial condition. When further increasing the current density up to 1,600 mA·g–1carbon, the lithium-oxygen cells with TiO2 NPs shows a discharge capacity of 800 mA·g–1carbon. Through these observations, we can conclude that the use of nanomaterials having catalytic activities and large surface area may be an effective way of expanding the specific capacity, enhancing rate capability and finally enabling the alleviation of cyclic degradation, even though if the material has semiconducting properties.
Conclusion
In summary, in order to search a suitable catalyst for lithium-oxygen batteries, the anatase TiO2 NPs with a diameter of 100 nm have been successfully synthesized by a simple hydrothermal method at 220℃ for 12 h reaction and processed for the cathode of lithium-oxygen cells. Thus, the prepared nanoparticles were tested in lithium-oxygen batteries and showed a remarkable high specific capacity of 9,200 mAh·g–1carbon with relatively stable cycle retention. The superior electrochemical performance and enhanced rate capabilities in lithium-oxygen batteries by using TiO2 nanoparticle is due to its efficient catalytic activity and large surface areas.