


1. Introduction
The energy derived from fossil fuels is expensive and causes environmental pollution owing to the emission of greenhouse gases and ultrafine powders during combustion. However, a major portion of the total energy in use is still obtained from conventional fossil fuels, despite vigorous research to develop several types of renewable energies for reducing the associated environmental threats. Additionally, electric vehicles (EVs) and energy storage systems (ESSs) are attracting considerable attention as highly energy-efficient equipment capable of reducing the use of fossil fuels. As a result, the importance of lithium ion batteries (LIBs) as one of the most prospective class of energy conversion and storage systems is increasing [1,2]. In response to this increasing demand, the market of LIBs has dynamically expanded from small portable electronic devices to large-sized EVs and ESSs. However, a remarkable improvement in the energy densities of LIBs is necessary to meet the power requirements for large-scale applications. High energy density in LIBs can be achieved by the application of a high operating voltage and the use of active materials with high theoretical capacity.
Researchers in the battery community have investigated various materials with high theoretical capacity, which can be categorized as Li-alloying (Si, Sn, Ge, etc.) and conversion type (CoO, FeO, NiO, etc.), as potential candidates for the negative electrode in LIBs [3-7]. These anode materials suffer from pulverization because of the large volume change during lithiation and delithiation. Many researchers have been attempting to achieve control over the morphology of the active materials and the physical characteristics of the binder to prevent this detrimental phenomenon [3,8]. Simultaneously, the electrolyte plays an important role in the formation of a stable passivation film on the electrode surface, whose area increases continually because of pulverization. Among several candidates, electrolytes with large amounts of fluoroethylene carbonate (FEC) additive have been studied to cover the newly exposed electrochemically active surfaces [9-11]. The electrolytes for conventional graphite anodes in LIBs usually contain a small quantity of additives, less than 10 vol%. However, the electrolytes for alloying-type anodes with high capacity require a large amount of additives such as FEC owing to the everincreasing surface area of the active materials resulting from pulverization [12,13].
On the other hand, it is difficult to identify the proper candidate material for the LIB cathode with high capacity because the complex host cannot accommodate a large number of lithium ions. Therefore, the researchers have been forced to search for other candidates operating at a high potential of over 4.2 V, so that there is a large potential gap between the anode and cathode. Among the various positive electrodes working at such a high potential, the spinel-type LiNi0.5Mn1.5O4 (LNMO) cathode has attracted much attention because of the high lithiation and delithiation potential of Ni2+/Ni4+ (~4.7 V vs. Li/Li+), low cost of Mn and Ni ions, and the robust crystalline structure resulting from the stabilization of Mn4+ by Ni2+ doping [14-16]. However, the high operating potential is beyond the electrochemical stability window of conventional electrolytes, and this restricts the applicability of the LNMO cathode [14,16,17]. The electrochemical stability window of conventional electrolytes is insufficient for them to resist decomposition on the surface of the LNMO cathode operating at high potentials. It is not an exaggeration to conclude that the severe oxidative decomposition of carbonate-based organic electrolytes at the surface is the largest hindrance to the use of the LNMO cathode. Generally, electrolyte decomposition results in the formation of resistive surface layers on the LNMO cathode, which consist of the decomposed products of lithium salts and organic carbonate solvents. Consequently, the LNMO cathode with traditional electrolytes suffers from continuous and irreversible capacity loss due to the detrimental electrolyte decomposition. Thus, one of the important challenges in the development of LIBs with the LNMO cathode is ensuring the presence of good passivation layers for maintaining high capacity over multiple cycles [14,18-21]. To relieve the resistance of the cathode materials during cycling, researchers have modified the electrolytes by introducing new types of lithium salts such as lithium difluoro(oxalate)borate, lithium tetrafluoroborate, and lithium 4-pyridyl trimethyl borate; new solvents such as fluorinated carbonates and phosphites; and various types of conventional additives for LiCoO2 cathode [14,22,23].
The surface layer, i.e., the solid-electrolyte interphase (SEI) plays a crucial role in resolving the oxidative decomposition of electrolytes, thereby affecting the cycle life characteristics of LIBs. Despite much investigation into the SEI on the negative electrodes, only a few studies have focused on improving the performance of the cathode-electrolyte interface layer, particularly for high-voltage cathode materials [24-26]. The main objective of this study is to confirm if the FEC-substituted electrolytes participate in the formation of SEI on the LNMO cathode. Herein, we selected the FEC additive as a substituted solvent, which exhibits an advantageous electrochemical performance for high-capacity anode materials such as Si and Sn. In particular, the presence of fluorine substituents in the electrolytes contributes to the increased oxidation stability in the high-voltage window. To demonstrate the physical and electrochemical characteristics of the FEC-oriented SEI on the high-voltage LNMO cathode, systematic analytical methods involving electrochemical, spectroscopic, and microscopic techniques were applied.
2. Experimental Section
Six different electrolytes with 1.0 M LiPF6 were prepared by varying the volumes of the three solvents: ethylene carbonate (EC, Panaxetec), diethylcarbonate (DEC, Panaxetec), and fluoroethylene carbonate (FEC, Panaxetec). The volumetric combinations of FEC, EC, and DEC were 0:5:5 (ref-electrolyte), 1:4:5 (FEC1-electrolyte), 2:3:5 (FEC2-electrolyte), 3:2:5 (FEC3-electrolyte), 4:1:5 (FEC4-electrolyte), and 5:0:5 (FEC5-electrolyte). The LNMO powders were prepared by the previously reported citric acid sol-gel method [16]. For electrochemical analyses, the composite electrodes were prepared by casting the slurry of the LNMO powder (active material), super-P (conducting agent, TIM-CAL), and polyvinyledene fluoride (binder, Solvay) in 94:3:3 and 80:10:10 wt% composition in N-methylpyrrolidinone (NMP; Sigma-Aldrich) solvent onto a piece of Al foil. The components of the slurry were thoroughly mixed using a mortar and pestle, before casting on the foil. Later, the slurry-casted electrodes were placed in a convection oven at 120┬░C to eliminate the NMP solvent. After pressing to enhance the interparticle contact and improve the adhesion to the Al current collector, the round electrodes, which have a loading level of 1 mA┬Ęh┬ĘcmŌłÆ2, were punched to have 1.1 cm diameter. The electrodes were carefully dried in a vacuum oven to eliminate the residual water. The electrochemical performances of the LNMO composite electrodes were evaluated using 2032-type coin cells assembled in an argon-filled glove box with lithium as the counter electrode, polypropylene as the separator (Celgard), and various electrolytes. Galvanostatic cycling measurements were performed using a Won-A-Tech WMPG-1000 battery cycler via repeated charging and discharging processes at various C-rates (1 C = 140 mA gŌłÆ1), in the voltage range of 3.5-4.9 V (vs. Li/Li+) at room temperature. For the 1st cycle, a current of 0.1 C was applied to reflect the formation process in the practical applications. For the 2nd and 3rd cycles, the charging and discharging currents were 0.5 and 0.2 C, respectively. Subsequently, charge and discharge sequences under 0.5 C were performed to evaluate the cyclability. For the self-discharge test, the cells were charged under a constant current of 0.1 C and a constant voltage of 4.9 V till a total capacity of 140 mA┬Ęh┬ĘgŌłÆ1 was attained. These fully charged cells were stored for five days at 120┬░C to observe the changes in the open circuit voltage (OCV). For field-emission scanning electron microscope (FE-SEM, Model JSM-7001F, JEOL) analysis, the cycled cells at the fully discharged state were disassembled in an argon-filled glove box to avoid air contamination, and the electrodes were thoroughly washed with dimethyl carbonate (DMC) to remove residual lithium salts and the solvent.
3. Results and Discussion
Fig. 1a displays the charge-discharge voltage profiles during the 1st cycle of the cells with the four electrolytes: ref-, FEC1-, FEC2-, and FEC3-electrolytes. Because of the large amount of FEC (greater than 40 vol%) in the electrolytes, the charge curves exhibited fluctuating potential at the end of charging (Fig. 1b). This trend reflects irreversible electrolyte decomposition, because the measured charge capacities were much higher than the theoretical capacity of the LNMO cathode, as calculated by lithium extraction. Unfortunately, this behavior restricts the use of the FEC4- and FEC5-electrolytes because the extremely low coulombic efficiencies cause rapid capacity decay owing to the loss of lithium ions in LIBs, forming a so-called rocking-chair system. Therefore, the amount of FEC in the electrolytes was limited to below 30 vol%, in order to evaluate the performance of the fabricated cells in commercial applications. From Fig. 1a, it was observed that every cell has one short and two long voltage plateaus at ~4.05, 4.76, and 4.72 V, which correspond to the electrochemical reactions of Mn3+/Mn4+, Ni3+/Ni4+, and Ni2+/Ni3+ respectively [15,16]. Theoretically, the components of the LNMO cathode have the initial oxidation states of Ni2+ and Mn4+. However, the pair of redox plateaus around 4.0 V indicated that the LNMO cathode has a small amount of Mn3+ impurity phase [17,27,28]. Despite the presence of this impurity phase, analysis of the electrochemical behavior under high potential conditions was not very critical because more than 90% of the charge and discharge capacities were recorded over 4.5 V (vs. Li/Li+).
Fig.┬Ā1.
Charge-discharge voltage profiles during the 1st cycle of the Li/LNMO (94:3:3 wt%) cells containing (a) ref, FEC1-, FEC2-, and FEC3-electrolytes, and the 1st (line) and 2nd (dotted) cycles of the Li/LNMO cells containing (b) FEC4- (upper) and FEC5-electrolytes (lower).
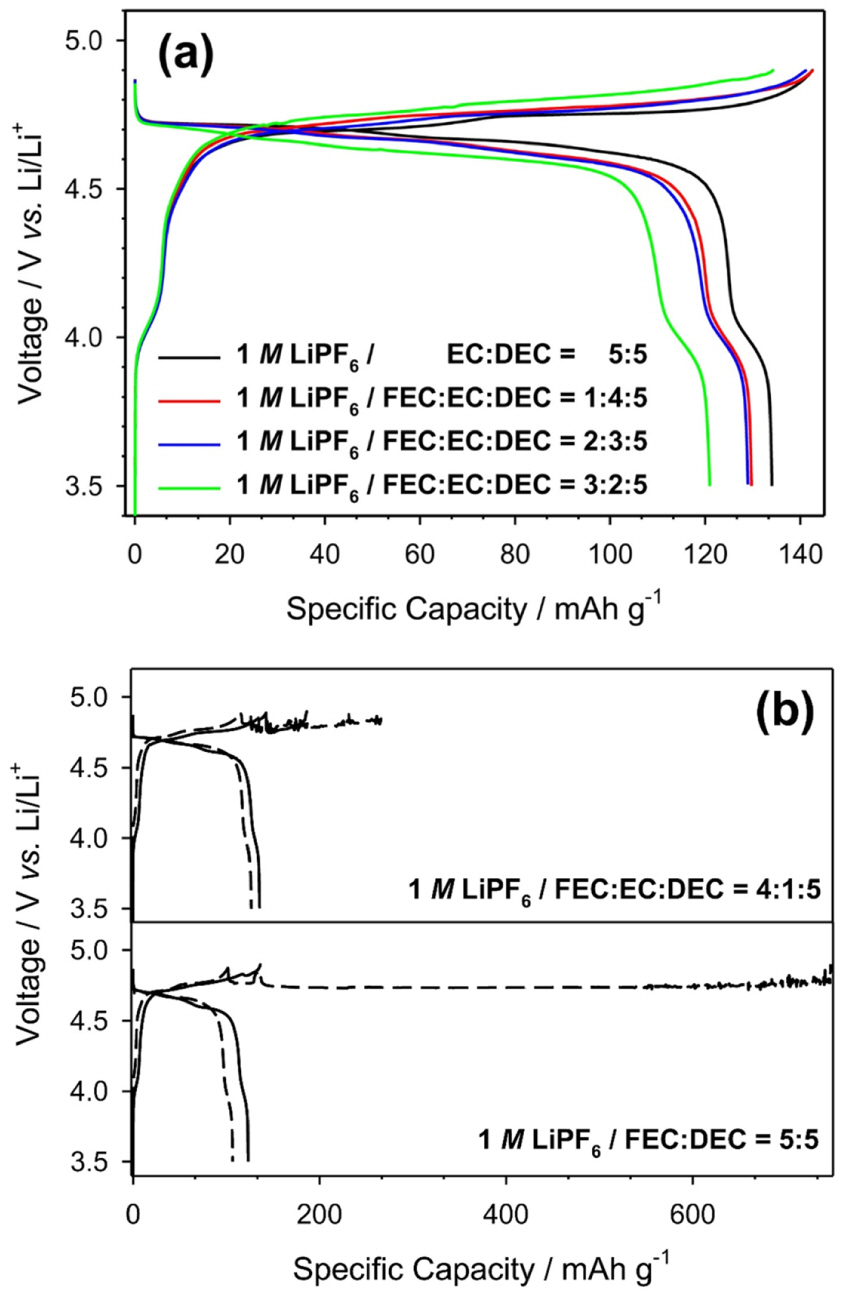
For the 1st cycle, the cells with ref-, FEC1-, and FEC2-electrolytes exhibited similar initial charge capacities of 142.6, 142.6, and 141.3 mA┬Ęh┬ĘgŌłÆ1 respectively, close to the theoretical capacity of the LNMO cathode (148.0 mA┬Ęh┬ĘgŌłÆ1). However, the cell containing the FEC3-electrolyte presented a relatively low charge capacity of 134.3 mA┬Ęh┬ĘgŌłÆ1 because of high polarization. In addition, the cell with the FEC3-electrolyte presented the lowest discharge capacity of 121.0 mA┬Ęh┬ĘgŌłÆ1. Similarly, the other two cells containing the FEC1 and FEC2 electrolytes showed relatively low discharge capacities of 129.8 and 129.0 mA┬Ęh┬ĘgŌłÆ1, respectively. On the other hand, the cell containing the ref-electrolyte showed a relatively high discharge capacity of 134.0 mA┬Ęh┬ĘgŌłÆ1, although these cells exhibited almost the same charge capacities after the 1st cycle. It was understood that the cells with the FEC1- and FEC2-electrolytes undergo more electrolyte decompositions during the charging process, even though they have similar initial charge capacities as that of the ref-electrolyte. Considering that the cells with the FEC4- and FEC5-electrolytes give exceptionally long plateaus, it is reasonable that FEC was electrochemically active on the LNMO composite electrode. Based on the polarized behavior of the cell containing the FEC3-electrolyte, it was thought that the electrochemical decomposition of FEC causes passivation of the LNMO cathode during the charging process.
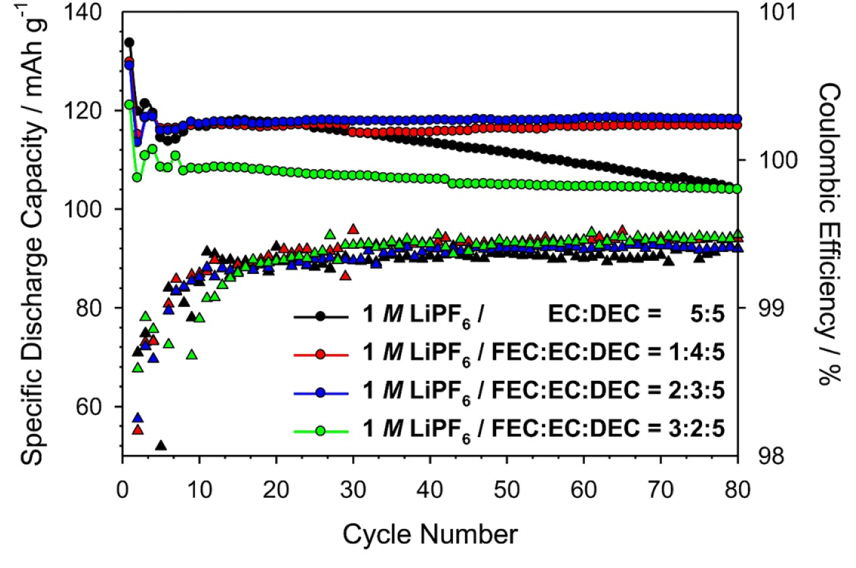
The cycle life and coulombic efficiency of the cells with the four types of electrolytes evaluated at 0.5 C after the 30th cycle, before which the formation process have been performed, are demonstrated in Fig. 2. At the initial cycling stage, i.e., during the first 20 cycles, the discharge capacities of the cells with the ref-, FEC1-, and FEC2-electrolytes were almost the same. However, after the 30th cycle, the cells containing the FEC1- and FEC2-electrolytes exhibited enhanced cyclability compared to the cell containing the ref-electrolyte. In addition, the cells containing the FEC1- and FEC2-electrolytes did not show any large capacity drop even after the 80th cycle. It was assumed that the surface of the LNMO cathode is gradually covered by the SEI because of the electrolyte decomposition. As the cycles proceeded, the more stabilized surface of the SEI resulted in increased coulombic efficiency. Therefore, the average coulombic efficiency of the cell with the ref-electrolyte during 80 cycles, except for the 1st cycle, was 99.27%. This value was than the less than the average values of the cells with the FEC1-, FEC2-, and FEC3-electrolytes (99.36%, 99.32%, and 99.34% respectively). This also supported the fact that the FEC-oriented SEI suppresses the continuous electrolyte decomposition on the LNMO cathode at high operating potential.
Fig.┬Ā2.
Comparison of the electrochemical cycling performances at 0.5 C. Cyclability (upper) and coulombic efficiency (lower) of the cells with ref-, FEC1-, FEC2-, and FEC3 electrolytes in the voltage range of 3.5-4.9 V (vs. Li/Li+) at room temperature.
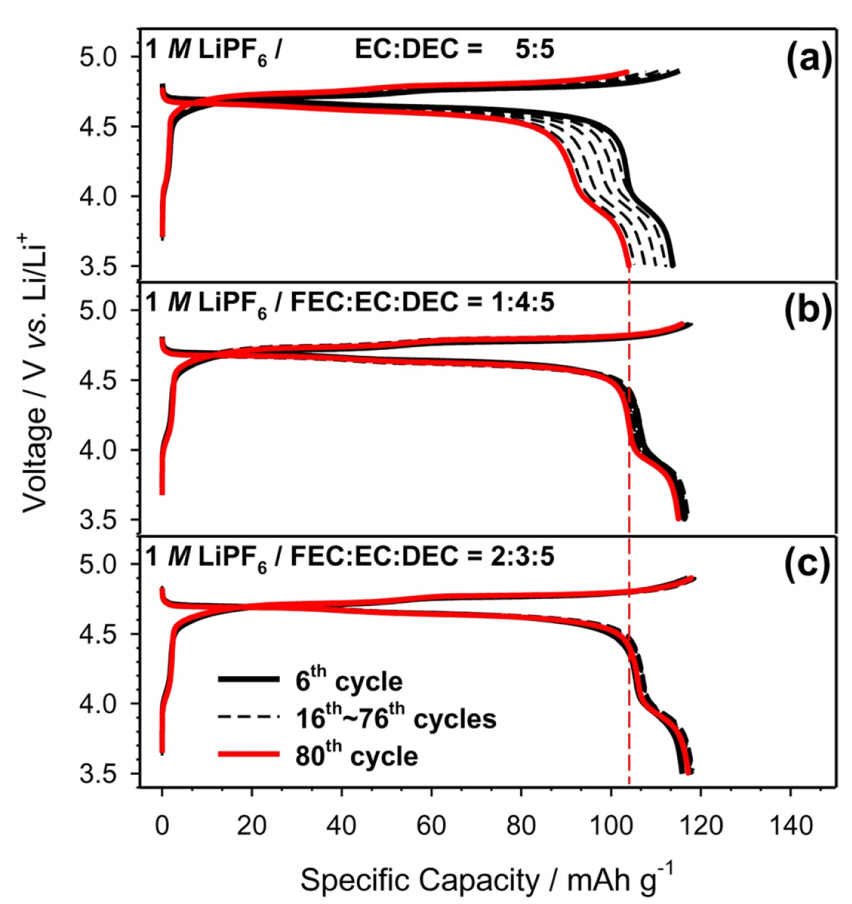
As shown in Fig. 3, the cells containing the FEC-substituted electrolytes were able to deliver higher reversible capacities compared to that containing the ref-electrolyte as the cycles proceeded. From Fig. 3a, it can be seen that the cell with the ref-electrolyte exhibited increasing polarization as the cycle number increased. Particularly, the diffusion polarization behavior after the 6th cycle became prominent at the end of the charge/discharge cycles because of the increased concentration polarization resulting from the resistance to lithium diffusion with repeated charging and discharging. In contrast, the voltage curves of the cells with the FEC1- and FEC2-electrolytes were successfully maintained due to the low kinetic hindrance. It was speculated that the surface failure from the irreversible capacities during cycling caused a high polarization, unlike the cases of the cells with the FEC-substituted electrolytes (Fig. 3b and c).
Fig.┬Ā3.
Selected charge-discharge voltage profiles. The cells containing (a) ref-, (b) FEC1-, and (c) FEC2-electrolytes were tested within the voltage range of 3.5-4.9 V (vs. Li/Li+) at room temperature.
To discuss the electrochemical behaviors in the cycling performances of the cells with the three types of electrolytes, FE-SEM analysis of the surface of the LNMO cathode was performed. Fig. 4a presents the SEM image of the pristine LNMO powders before cycling, indicating that the LNMO particles have sharp edges because of the highly ordered crystalline spinel structure. Fig. 4b shows the SEM image of the cell containing the ref-electrolyte after the 100th cycle, which reveals the continued presence of the sharp edges of the LNMO particles. Figs. 4c and d demonstrate the thin surface film deposit from the other two cells containing the FEC-substituted electrolytes after 100 cycles. The additional surface films are distinct in Figs. 4c and d, indicating that the addition of FEC to the electrolytes favors the formation of the surface film, as mentioned previously.
Fig.┬Ā4.
FE-SEM images of the surface of the LNMO (94:3:3 wt%) cathode with the three electrolytes: (a) ref-electrolyte before cycling, (b) ref-, (c) FEC1-, and (d) FEC2-electrolytes after 100 cycles.

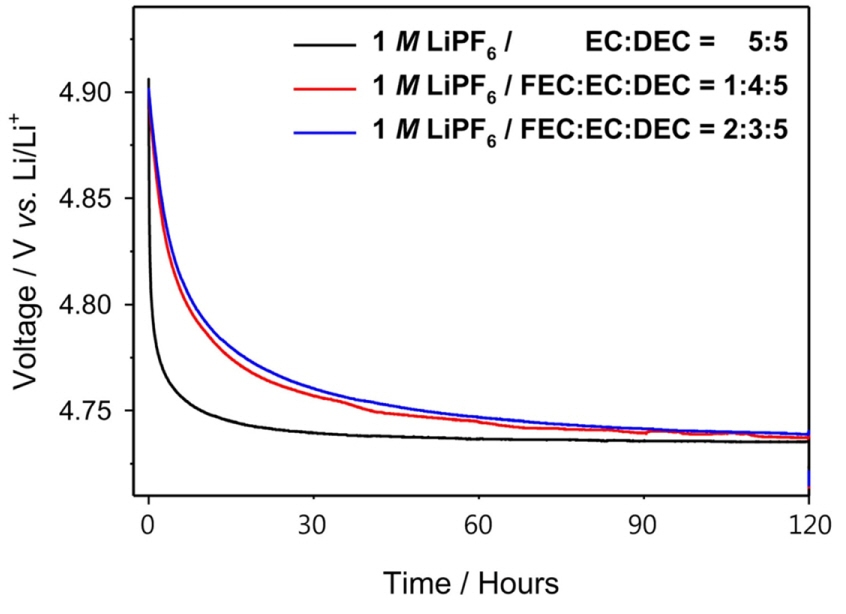
Fig. 5 shows the shelf life characteristics of the fully charged cells that were controlled to have the same charge capacity of 140 mA┬Ęh┬ĘgŌłÆ1. At this point, the large voltage drop was generated from undesired side reactions such as the transport of electrons from the electrolyte to the cathode without passing through an external circuit. The details of the self-discharge mechanism can be described as below.
Fig.┬Ā5.
Discharge curves of the cells with ref-, FEC1-, and FEC2-electrolytes in the open-circuit state for five days at 120Ōäā.
Electrolyte ŌåÆ eŌłÆ + Electrolyte+ + by-products
LixNi0.5Mn1.5O2 + yLi+ + yeŌłÆ ŌåÆ Lix+yNi0.5Mn1.5O2
The cell containing the ref-electrolyte showed a more rapid voltage drop between 4.90 V and 4.75 V as compared to the other two cells containing the FEC-substituted electrolytes, indicating that the side reactions were suppressed by the FEC-oriented SEI. The open-circuit potential of the LNMO cathode containing Ni4+ was approximately 4.75 V (vs. Li/Li+) [29]. Below 4.75 V, the potential changes were more rapid because of the large number of electrons stored in the double layer. During this period, even a slight progress of the electrochemical side reactions caused a large voltage drop. Additionally, the potential of the cell with the ref-electrolyte stabilized at 4.75 V, where the Ni3+/Ni4+ redox reaction occurs; however, it was assumed that the electrons were still being consumed by the electrochemical side reactions. The voltage curves resulting from the electrochemical side reactions indicate that the FEC-oriented SEI successfully suppresses the side reactions between the surface of the LNMO electrode and the electrolyte.
Fig. 6 shows the Nyquist plot of the fully charged LNMO electrode with the ref- and FEC1-electrolytes. Two semi-circles, whose diameters reflect the surface film resistance (Rfilm) and the charge transfer resistance (Rct), were distinctly observed in both the plots. The semi-circle in the high-frequency domain (>125.89 Hz) represents Rfilm, while the other represents Rct, as an equivalent circuit shown in the inset in Fig. 7. At the fully charged state, the total resistance of the LNMO cathode with the refelectrolyte was higher than that of the LNMO cathode with the FEC1-electrolyte. The reduction of the resistance supports the enhanced cyclability observed in Fig. 2.
Fig.┬Ā6.
Nyquist plots of the fully charged three-electrode cells with ref- and FEC1-electrolytes during the 1st cycle.
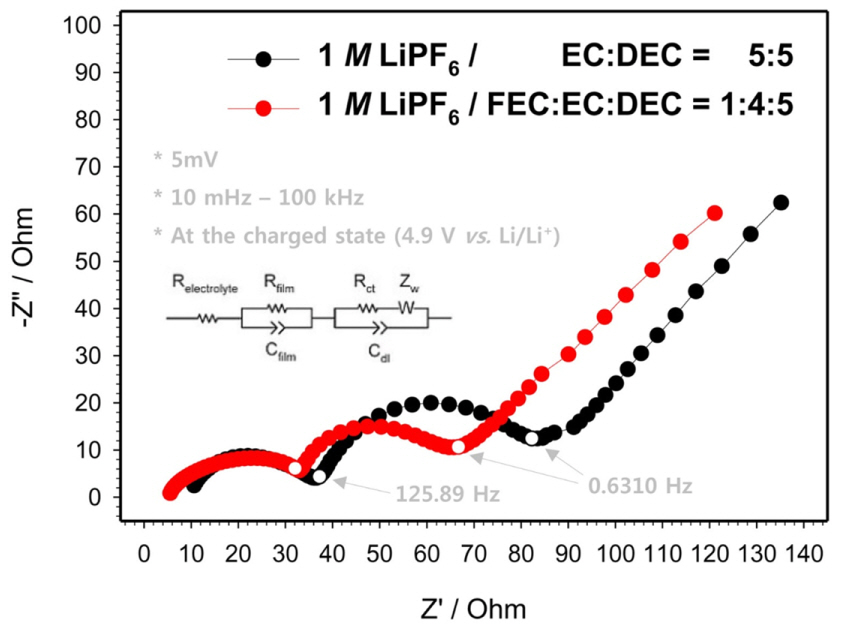
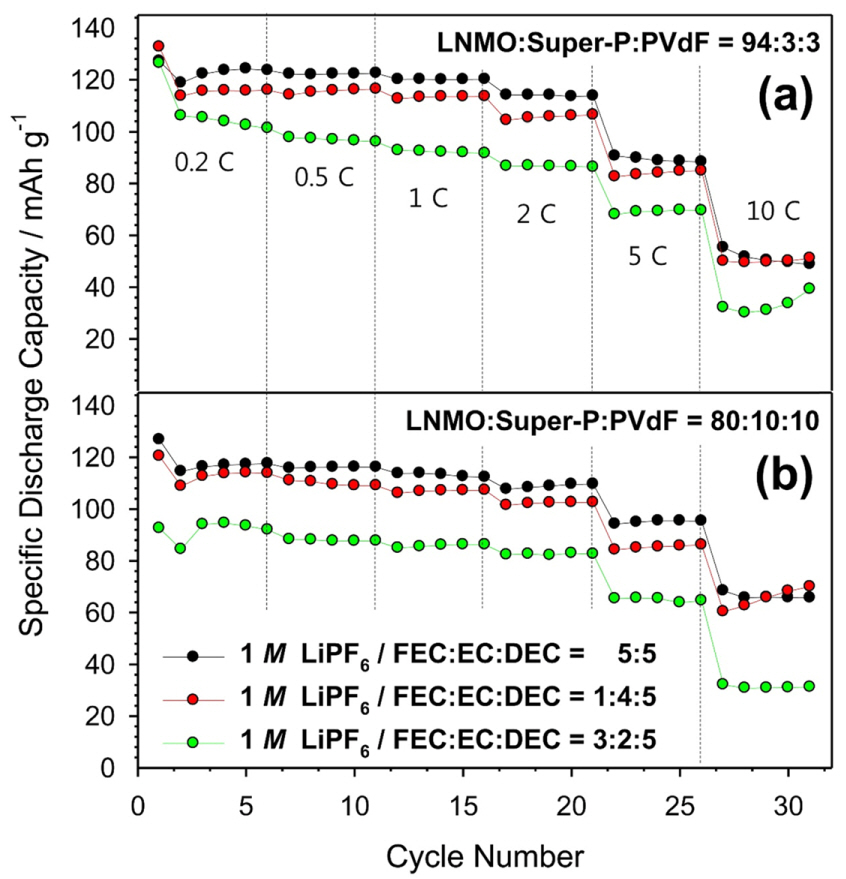
Fig. 7 compares the rate capabilities of the cells with the two types of electrode compositions at various current densities during the discharge process. With an increase in the current, the polarization due to several kinetic hindrances such as the ohmic resistance, ion-diffusion limitation, and charge transfer resistance had a negative effect on the maintained high capacity. As seen in Fig. 7a, the cells containing the FEC-substituted electrolytes exhibited poorer rate capabilities than the cell containing the ref-electrolyte. The rate capabilities of the LNMO electrode (94:3:3 wt%) correlated well with the voltage curves in Fig. 1. As shown in Fig. 7b, the cells with the FEC-substituted electrolytes and the LNMO electrode (80:10:10 wt%), which contains more carbon compared to the other electrode composition (94:3:3 wt%), also exhibited poor rate capabilities. It was understood that the increased resistance of the LNMO electrode due to the FEC-oriented SEI cannot be resolved by increasing the carbon content to ensure the electron-pathway between LNMO particles and conducting agents in the electrode. Given that the carbon additive in the electrode could not relieve the diffusion resistance, the electronic insulation due to the SEI is not the main factor limiting the rate capability. Instead, the solid diffusion in the SEI is the main factor limiting the rate capability.
4. Conclusions
We have demonstrated an advanced methodology wherein EC is substituted with FEC in the EC/DEC-based electrolyte for the cells with the high-voltage LNMO cathode to enhance the electrochemical performance. The cycling tests demonstrated that the FEC-oriented SEI alleviates the capacity fading and suppresses the side reactions during cycling. However, the high polarization resulting from the formation of the FEC-oriented SEI is observed in the cells containing the FEC-substituted electrolytes. Despite the improved cycle life, the rate performance of the LNMO cathode with the FEC-substituted electrolytes is inferior to that of the LNMO cathode with the ref-electrolyte, even after increasing the carbon content in the electrode. This attempt to substitute some solvents in the traditional electrolytes with FEC may be a feasible approach for achieving a progressive LIB system in a high voltage window.




