


1. Introduction
In recent years, the threat of global warming and the emerging awareness of the increasing atmospheric CO2 concentrations have placed a spotlight on carbon capture, storage, and utilization (CCSU) [1,2]. CCSU technologies are garnering considerable attention as they allow for the conversion of CO2 to useful chemicals, thereby decreasing the atmospheric concentration of CO2 substantially [3-5]. CO2 is an attractive building block for C1-chemistry; however, the conversion of CO2 is quite challenging due to its inert and stable nature. Several methods for the activation and conversion of CO2 have been proposed, such as chemical [5], biological [6], photochemical [7,8], and electrochemical methods [9-12]. Among these technologies, the electrochemical method is the most promising and benign, owing to its convenient operation and ability to control the yield and selectivity of the produced chemicals.
Metallic catalysts have mainly been investigated for electrochemical CO2 conversion. Various lowcarbon species are generated during this process, e.g., HCOOH [11,13-15], CH4 [9,16], CO [17-19], and C2H4 [20]. When a simultaneous hydrogen evolution reaction (HER) occurs, CO/H2 mixture (syngas) can be produced in a single electrochemical cell [11,14]. This strategy has drawn great attention because the syngas can be utilized and transformed into other valuable chemicals such as methanol and ethanol via the Fischer-Tropsch process. In all previous studies, each metal was found to show exclusive selectivity for a converted product. For example, the d-block transition metals such as Au, Ag, and Co were effective in converting CO2 to CO, while the p-block transition metals such as Sn and In could actively produce HCOOH [21]. In addition, Zn, Ni, and Cu, which are attractive candidates due to their abundance and low cost, were found to selectively generate CO, H2, and CH4, respectively [21]. Unfortunately, the conventional metal catalysts normally require high energies for the activation of CO2, and therefore, exhibit low energy efficiency. Moreover, the metal catalysts, whether planar or covalently bound to supports, possess limited electrochemically active surface areas. In terms of kinetics, the rate of the electrochemical reaction is governed by the rate of the catalytic reaction and the areal concentration of the active catalyst. Therefore, in addition to the modification of such metal catalysts, it is necessary to explore other types of catalysts for the electrochemical conversion of CO2. Notably, there have also been reports on the use of enzymes as catalysts toward the electrolytic conversion of CO2. However, the reaction conditions for the enzymatic conversion of CO2 should be restricted to neutral pH and mild temperatures, as otherwise, the enzymes would easily become unstable and inactive [22-24].
Recently, there have been attempts to apply metal organic frameworks (MOFs) as catalysts for the electrochemical conversion of CO2 [25-28] and H2 production [29,30]. MOFs are made of metal ions coordinated to organic ligands, giving rise to characteristic three-dimensionally ordered crystalline structures. MOFs are highly porous, thermally stable, and chemically functional. In addition, MOFs can uniformly retain active metal nanoparticles at high densities within their structure, and therefore, increase the amount of surface-bound catalyst, resulting in a large active surface area. These extraordinary properties of MOFs are attractive for use in the electrochemical conversion of CO2 and production of H2. Previous studies focused on different types of MOF structures such as porphyrin-based [25,26], benzenetricarboxylate (BTC)-based [27], and rubeanate-based MOFs [28] for CO2 reduction. Even though some samples showed promising activity, the previously suggested MOF structures required long and multiple steps for synthesis.
Among the current MOF materials, MOF-74 is known to exhibit an exceptionally large surface area and higher CO2 adsorption capacity under atmospheric conditions compared to the other MOFs [31,32]. In particular, M-MOF-74 (M = transition metal) is designated for isostructural compounds with the composition M2(dhtp)(H2O)2·8H2O (dhtp = 2,5-dihydroxyterephthalate), in which the metal species act as vertices of the hexagons connected by organic linkers [33]. Because the M-MOF-74 structure exhibits Lewis acidic sites (open metal sites) upon heating in vacuum, the CO2 molecules, which act as Lewis bases, strongly bind with the open metal sites [33]. In addition, M-MOF-74 can be easily obtained because its synthesis is relatively simple and requires mild condition, which in turn makes it possible to change the central metal impregnated in MOF-74. In fact, the present synthetic process for M-MOF-74 involves only heating, mixing, and cooling. This is more facile than other methods such as electrophoretic deposition [25], atomic layer deposition [26], and electrochemical deposition [27], which require complex apparatus such as a sealed chamber or a three-electrode electrochemical cell. More importantly, the representative MOF-74 can retain its original structure during the catalysis or even after any further treatment. To the best of our knowledge, M-MOF-74 has not yet been explored as a catalyst for electrochemical CO2 conversion. Therefore, M-MOF-74 can be a reliable catalyst for CO2 conversion among the other MOFs and help us investigate the effect of the central metal on the catalysis.
In this work, we attempted aqueous electrochemical reduction of CO2 on the M-MOF-74 (M = Co, Ni, Zn) catalyst, using a proton exchange membrane (PEM)-based electrolyzer. The synthesis of the MOF-74 catalyst is relatively facile as compared to the synthesis of other types of catalysts because it just requires simple mixing, heating, and cooling for structuring. Hence, M-MOF-74 with various central metals can be obtained via an inexpensive and high-yield process. Thus far, only Fe [25], Co [26], and Cu [27,28] have been investigated as the central metal in the MOF structure for the electrochemical conversion of CO2, regardless of the type of MOF. From this experiment, we expect to demonstrate the effect of the type of central metal in MOF-74 on the electrochemical conversion of CO2, in terms of the reduced product and conversion efficiency.
2. Experimental Section
2.1. Synthesis of M-MOF-74
All the chemicals were purchased from Sigma-Aldrich (Seoul, Korea) and used as received. Ni- and Co-MOF-74 were synthesized by a modification of the literature procedure [34]. For Ni-MOF-74, a mixture of 2,5-dihydroxyterephthalic acid (H4DOBDC, 0.478 g, 2.41 mmol) and Ni(NO3)2·6H2O (2.378 g, 8.18 mmol) was added to a mixed solvent of N,N-dimethyformamide (DMF)-ethanol-deionized water (1:1:1, v/v/v%, 200 mL). The suspension was uniformly mixed by ultrasonic stirring. A vial containing the suspension was placed in an oven at 100°C for 24 h, followed by cooling to room temperature. The supernatant was separated from the precipitate and replaced with methanol. The precipitate was rinsed with methanol four times in two days. The remaining solvent was removed under vacuum at 250°C for over 5 h, leaving a brownish-yellow crystalline powder, Ni-MOF-74. The final product was stored under vacuum to avoid contamination. The synthetic procedure for Co-MOF-74 is almost the same as that described above, except for the composition of the mixture: H4DOBDC (0.482 g, 2.43 mmol) and Co(NH3)2·6H2O (2.377 g, 8.67 mmol). A dark red-purple powder, Co-MOF-74, was obtained and vacuum stored.
Zn-MOF-74 was synthesized and activated according to a method described in literature [35]. H4DOBC (2.5 g, 13 mmol) and Zn(NO3)2·4H2O (10.0 g, 38 mmol) were dissolved in 500 mL of DMF with stirring, followed by the addition of 25 mL of deionized water. The mixture was heated in an oven at 100°C for 20 h. After decanting the supernatant, the product was rinsed with DMF and immersed in methanol for 6 days, during which time the activation solvent was decanted and replenished three times. The solvent was finally removed under vacuum at 270°C, yielding the porous material, Zn-MOF-74.
2.2. Material Characterization
The powder X-ray diffraction (PXRD) patterns of M-MOF-74 were recorded by a diffractometer (XDS 2000, Rigaku, Texas USA) using Ni-filtered Cu Kα radiation (λ = 1.5412 Å) over the range 3° < 2θ < 50° in 0.1° steps, with a counting time of 1 s per step. The morphologies and shapes of the M-MOF-74 samples were obtained by using a transmission electron microscope (TEM, F20, Tecnai).
N2 adsorption/desorption isotherms were measured volumetrically at 77 K over the range 7.0 × 10−6 ≤ P/P0 ≤ 1.00 by an Autosorb-iQ gas sorption analyzer outfitted with a micropore option from Quantachrome Instruments (Boynton Beach, Florida, USA) using the Autosorb-iQ Win software package. After solvent exchange of the as-synthesized materials with MeOH (2 × 10 mL, 12 h each time), the samples were activated (i.e., degassed) at 250°C for 5 h using the outgas port of the Autosorb-iQ instrument. The specific surface areas available for N2 adsorption were calculated using the Brunauer-Emmett-Teller (BET) model in the linear range, as determined using the consistency criteria.
2.3. Electrochemical Characterization with Gas Analysis
We have developed a laboratory-made PEM-embedded electrolyzer, where the cathodic and anodic compartments are separated by a polymer membrane (NRE-211, 25.4 μm, Dupont™). The specification of the three-electrode system is as described here. A catalyst-coated rotating disk electrode (RDE, 0.126 cm2) with a glassy carbon tip was used as the cathode. A saturated calomel electrode (−0.241 V vs. standard hydrogen electrode) and a Pt mesh (Pt thickness = 100 μm, 4 cm2) were used as the reference and counter electrodes, respectively.
A potentiostat (PGSTAT302N, Autolab) was used in all the electrochemical measurements. The electrolyte, 0.5 M KHCO3, was de-aerated with N2 gas for 30 min and saturated with CO2 via purging at a flow rate of 30 mL min−1 for at least 30 min. The electrochemical reduction potential of CO2 was investigated by performing linear sweep voltammetry (LSV) from 0.0 to −1.2 VSCE at a sweep rate of 10 mV s−1. The time-dependent current decay upon the application of a step potential was measured by chronoamperometry at −1.6 VSCE (overpotential η = −0.8 V) for 60 min. All the potentials in this study are expressed with reference to the reversible hydrogen electrode. The catalytic activity was measured based on the reduction current during the experiment and the concentration of the produced CO and H2 determined from the gas analysis.
The concentration of the product gas was measured by real-time gas chromatography (GC, 7890B, Agilent, Santa Clara, USA). The gas was directly delivered to the sampling loop of the GC system in a stream of CO2. CO2 was continuously supplied into the catholyte at 10 ccm, controlled by the mass flow controller. The dual-detector (a thermal conductivity detector, TCD and a methanizer-combined flame ionization detector, methanizer-FID)-embedded GC system can analyze both permanent and organic gases. The apparatus used for the electrochemical reduction of CO2 and successive analysis of the product gases is depicted in Fig. 1.
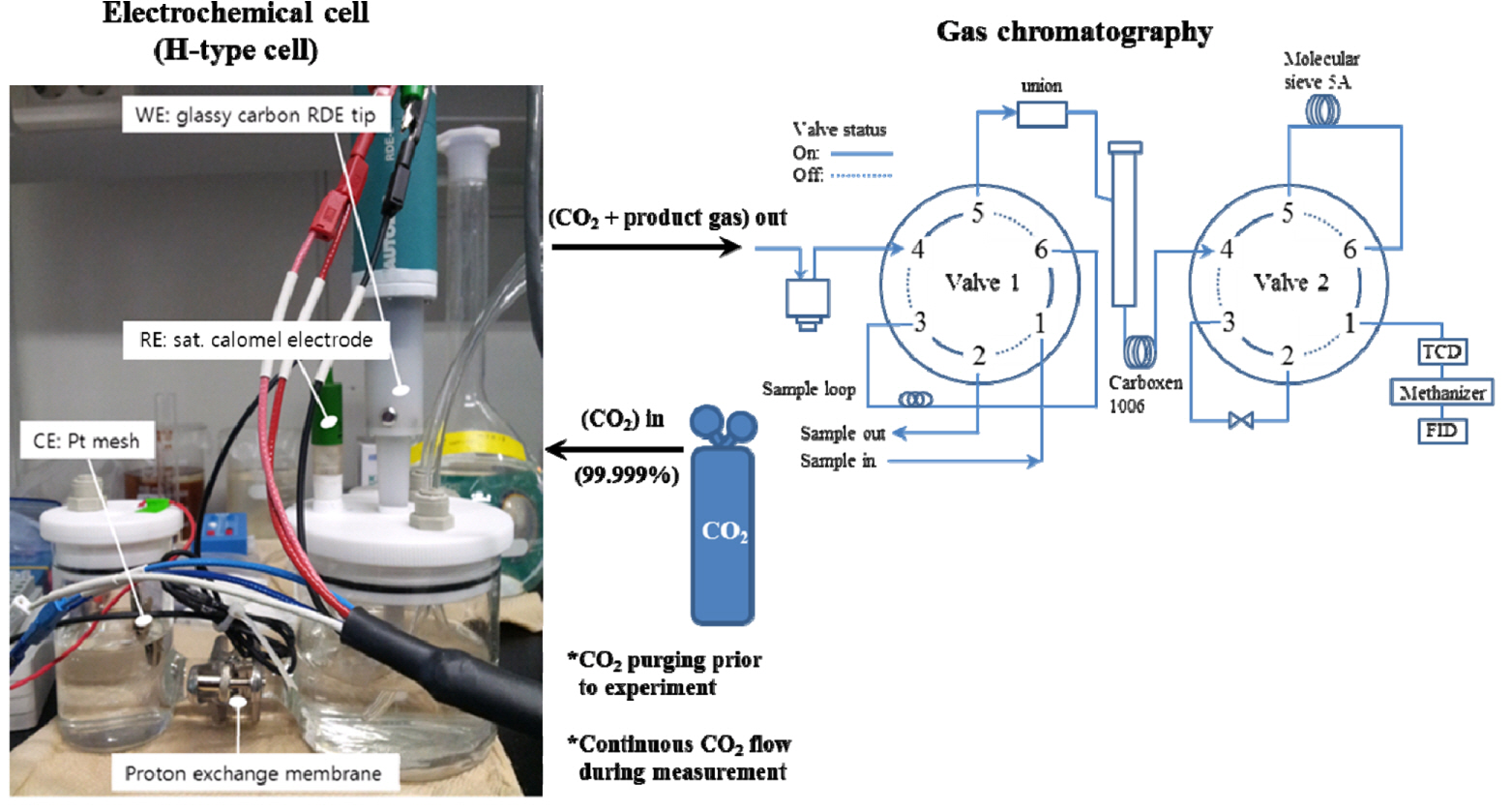
3. Results and Discussion
The MOFs, i.e., Co-, Ni-, and Zn-MOF-74, were prepared and purified according to a reported procedure [34,35]. Fig. 2(a) shows that the PXRD patterns of the as-synthesized samples matched well with the literature data and simulated patterns, indicating successful synthesis of the desired MOFs. The microporosities of the samples were measured via N2 adsorption-desorption isotherms. As depicted in Figure 2(b), all the isotherms exhibited type-I behavior without hysteresis, which implied a microporous structure. Moreover, the isotherms were found to quickly rise at the low-pressure region (P/P0 < 0.1) indicating that the open metal sites are easily accessible to the gas molecules. The BET surface areas of the degassed Co-, Ni-, and Zn-MOF-74 were 1356, 1239, and 1134 m2 g−1, respectively, which were in excellent agreement with the reported M-MOF-74 values [34]. Because the surface area is reported as area per unit mass, the material with a lighter central metal had a larger surface area.
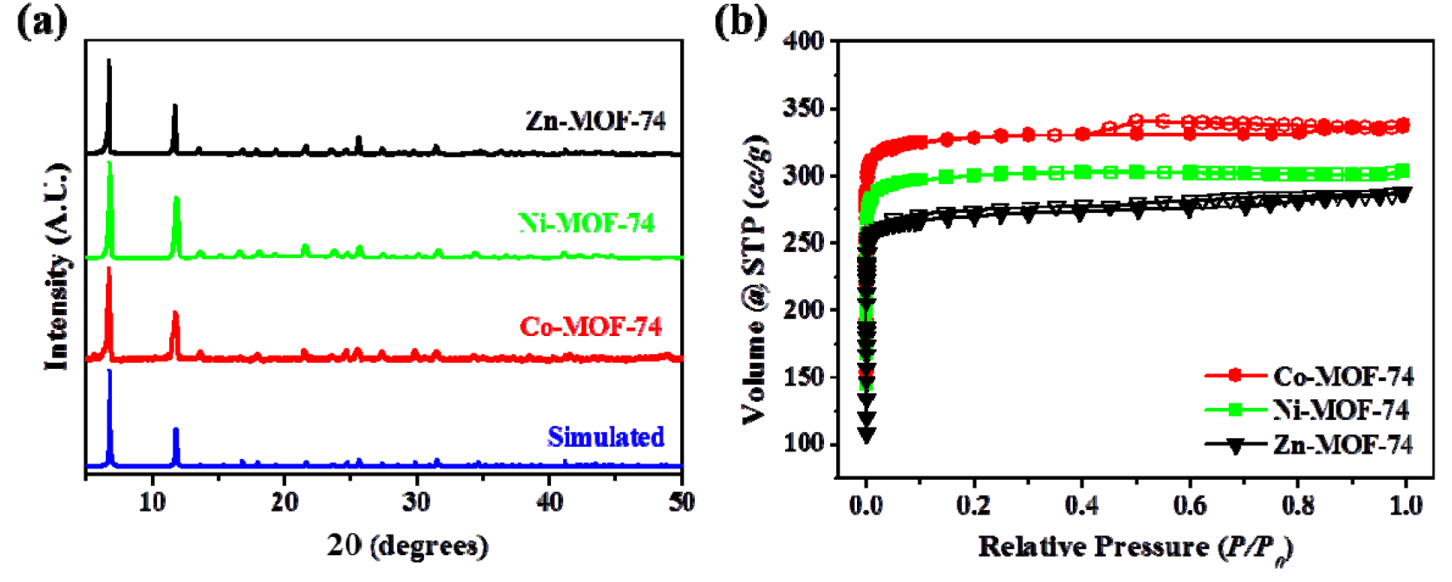
Fig. 3 shows the electron microscopic images of M-MOF-74. It appeared that the frameworks possessed polygonal shapes. From the TEM-EDX analysis, it was found that Co-MOF-74, with a size of ~0.3 μm, contained 15.2 wt% Co uniformly distributed throughout the framework. The size of the individual Co nanoparticles was ~1.93 (± 0.21) nm. Ni-MOF-74 contained 16.7 wt% Ni, whose individual particle size was ~1.48 (± 0.14) nm. Zn-MOF-74 had the highest metal content among the M-MOFs (19.4 wt%), although the individual Zn nanoparticles were not clearly observed. Generally, the metal loading in the MOF structure was limited to a few wt% because of restricted space [36]. The observed high metal loading in M-MOF-74 makes it increasingly suitable for the role of an electro-catalyst, in which the loading is normally 20-30 wt%. It is to be noted that TEM-EDX can provide similar quantitative results as ICP-MS, as reported previously [37,38].

Fig. 4 demonstrates the electrochemical response of M-MOF-74, as measured by LSV in the potential range of 0 to -1.2 VRHE in 0.5 M KHCO3 (aq). The current response with voltage was reproducible, indicating that the structure of the framework is stable. A clear increase in the reduction current was observed for all the M-MOF-74 samples as the potential was swept in the negative direction. Because the electrolyte was fully saturated with CO2, the reduction current was responsible for both the electrochemical reduction of CO2 and H2 production. The reduction current of Co-MOF-74 and Ni-MOF-74 started increasing at −0.25 and −0.56 VRHE, respectively. However, the on-set potential of Zn-MOF-74 was shifted to ~-0.65 VRHE. Given that the hydrogen evolution reaction (E° = 0.0 VRHE) precedes the reduction of CO2 (E° = −0.11 VRHE), it is more probable that the initial current response was due to the hydrogen evolution. It is known that Co and Ni are efficient hydrogen evolution catalysts, and they have been intensively investigated [39–41]. Hence, the smaller overpotentials for Co- and Ni-MOF-74 compared to Zn-MOF-74 were ascribed to their superior catalytic activities toward hydrogen evolution. Meanwhile, the shift of the on-set potential could also be rationalized in terms of the activity toward CO2 reduction, because Zn required a higher potential than Co and Ni to electrochemically reduce CO2, as reported by Y. Hori [42] and K. Hara [43].
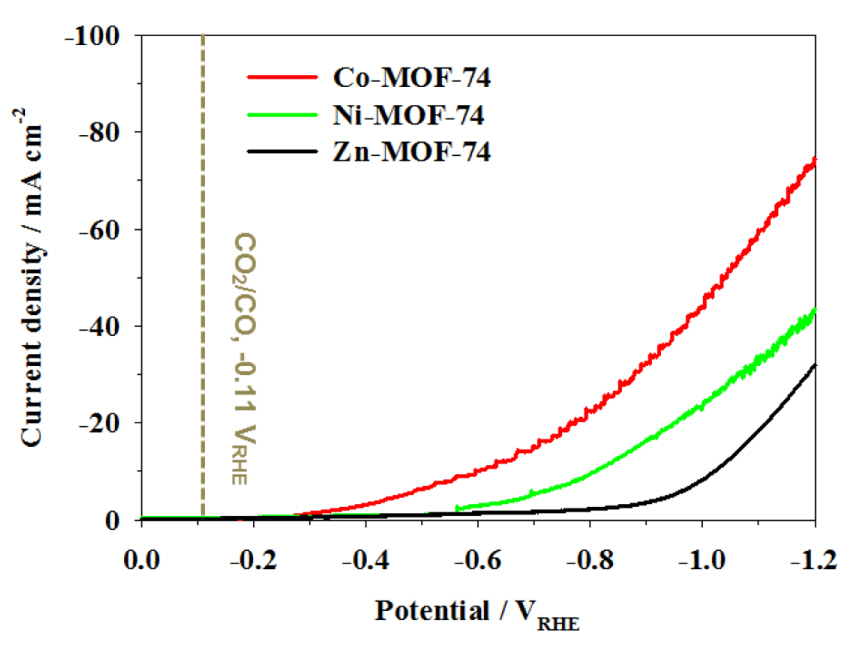
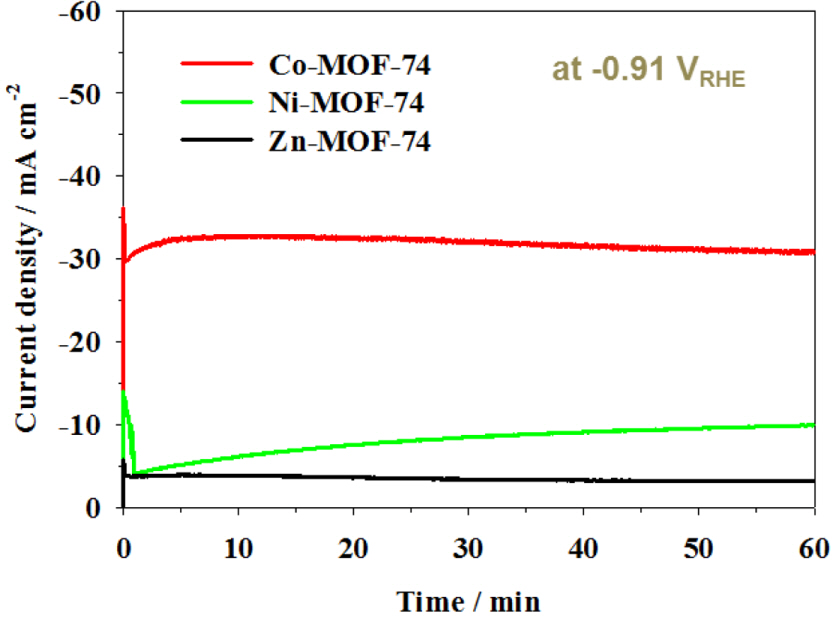
Fig. 5 presents the time-dependent electrochemical activity and stability of M-MOF-74 catalysts, evaluated through chronoamperometric measurements for 60 min at a controlled potential of −0.91 VRHE. The abrupt current decrease observed in the chronoamperometry experiments was attributed to the double-layer charging current [44,45]. The geo metric current density of Co- and Zn-MOF-74 reached a stable state a few minutes after applying the potential. The current density of Ni, however, gradually increased with time, probably due to slow activation of the Ni particles bound to the framework, and became stable with time. All the steady-state current densities matched well with the values obtained from LSV, as shown in Figure 4. The applied potential was determined based on the fact that the current density, greater than 5 mA cm−2, seemed to be academically meaningful as evidenced by the previous study [42].
Figs. 6(a-c) provide the partial current densities of CO2 and H2 reduction on M-MOF-74 at −0.91 VRHE as a function of electrolysis time. The partial current densities (j) were calculated from the comprehensive product gas analysis and the electrochemical activity [46]. Co-MOF-74 exhibited superior selectivity toward H2 reduction (jH2) over CO2 reduction (jCO2). Likewise, Ni-MOF-74 generated a large quantity of H2 at the given potential, and showed a low jCO2. This suggested that between the competitive reactions of H2 evolution and CO2 conversion, the former prevailed on both Co- and Ni-MOF-74. It can be assumed that Co-/Ni-MOF-74 would be an effective H2-evolving catalyst in the electrolysis of water. In the case of CO2 reduction on both the catalysts, the current initially increased but gradually decreased with electrolysis time, indicating that Co-/Ni-MOF-74 possessed low durability for CO2 reduction. On the other hand, Zn-MOF-74 presented a much greater jCO2 compared to the others, indicating that Zn-MOF-74 could substantially reduce CO2. It is also notable that jCO2 was maintained almost constant during the electrolysis time.
Fig. 6.
Partial current density for H2 production (triangles), CO2 reduction (solid squares), and CO production in CO2 reduction (open squares) on (a) Co-MOF-74, (b) Ni-MOF-74, and (c) Zn-MOF-74 at −0.91 VRHE for 60 min.
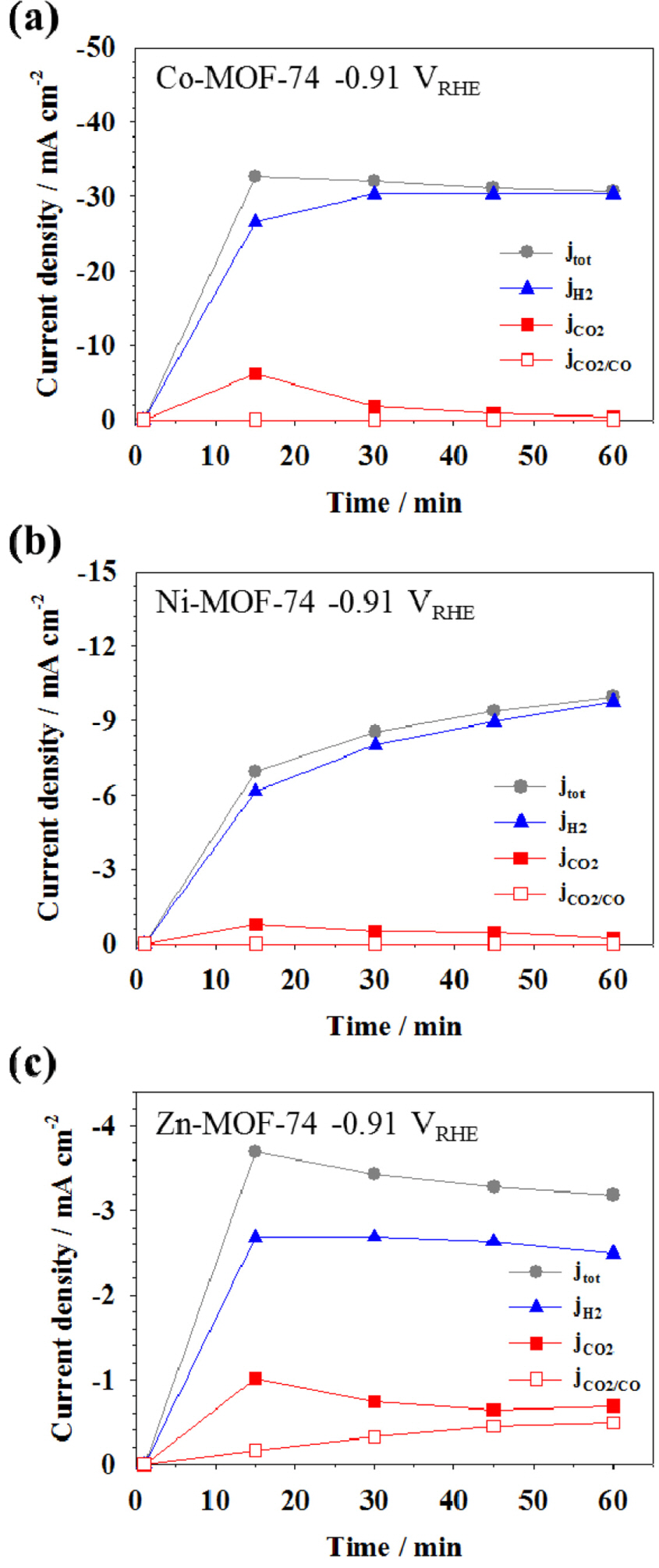
The fact that no CO was detected from the GC analysis of Co-/Ni-MOF-74 and jCO2/CO remained zero suggested that the jCO2 was related to the conversion of CO2 to compounds other than CO. These compounds were not detectable by GC; therefore, it was assumed that they were in liquid state. It was earlier reported that Co and Ni could convert CO2 into formate with relatively low efficiency [42]. Therefore, we could tentatively conclude that the liquid component would be formic acid. Unlike Co- and Ni-MOF-74, Zn-MOF-74 produced a notable amount of CO. This suggested that Zn-MOF-74 shows higher CO selectivity than the other types of M-MOFs, in the conversion of CO2.
Fig. 7 shows the relative proportion of the reduced products when the electrolysis proceeded for 60 min. It was concluded that Co-/Ni-MOF-74 could electrochemically convert a small quantity of CO2, and produce a large quantity of H2. Meanwhile, Zn-MOF-74 led to 21.72% CO2 conversion and 78.28% H2 production. The CO selectivity obtained from Zn-MOF-74 (15.40%) was greater than that reported for a Cu rubeanate-based MOF (2%) [28] but smaller than that reported for an Fe porphyrin-based MOF (50%) [25]. However, the CO productivity (−0.69 mA cm−2) of CO partial current was higher than that of Cu rubeanate-based (−0.36 mA cm−2) and Fe-porphyrin-based MOFs (−0.65 mA cm−2) at the same reduction potential. From this result, we could conclude that the electrocatalytic activity of Zn-MOF-74 is more promising than the other MOFs. Moreover, the simultaneous production of CO + H2 (syngas) on Zn-MOF-74 would definitely lead to the production of a valueadded fuel via a gas-to-liquid reaction, thus confirming the potential use of this material as an electrocatalyst in the selective conversion of CO2.

4. Conclusions
In this study, we have demonstrated the applicability of the M-MOF-74 (M = Co, Ni, Zn) catalyst for the electrochemical reduction of CO2. The as-synthesized M-MOF-74 possessed a microporous structure and contained uniformly distributed metal nanoparticles in its interior. According to the BET measurements, all the M-MOF-74 catalysts had a surface area greater than 1134 m2 g−1. The metal content of M-MOF-74 varied from 15 to 20 wt%. The results of the study demonstrated that M-MOF-74 showed different electrochemical activities toward the reduction of CO2 and production of H2. Smaller overpotentials were observed for the reactions with Co- and Ni-MOF-74 than for those with Zn-MOF-74, owing to the superior catalytic activities of the former catalysts toward H2 evolution. It was revealed that the reduced products and their selectivity strongly depended on the type of central metal. Co- and Ni-MOF-74 presented superior selectivity toward H2 reduction over CO2 reduction. On the other hand, Zn-MOF-74 could produce both CO and H2 with selectivities of 15.4% and 78.28%, respectively, suggesting that it could be a potential heterogeneous catalyst for syngas production via the electrochemical conversion of CO2. In this preliminary attempt to apply M-MOF-74 as a catalyst for the electrochemical reduction of CO2, the improvement in the conversion efficiency and the kinetics fell short of our expectation. Based on the results, however, it would be of great interest to fabricate binary metal catalysts impregnated in MOF-74, i.e., CoZn-MOF-74 or NiZn-MOF-74, and use them for the production of high yield syngas in future research.




