1. Introduction
As the technology converting CO2 to formic acid advances, the researches toward direct formic acid fuel cell (DFAFC) becomes more important for portable devices and power sources at off-grid locations [1–5].
Pt-based electrochemical catalysts have been intensively investigated toward formic acid oxidation (FAO) [6–11]. FAO is known to proceed in dual path mechanism [12–16]: dehydrogenation path (HCOOH → CO2 + 2H+ + 2e), and dehydration path (HCOOH → H2O + poisonous CO; poisonous CO + H2O → CO2 + 2H+ + 2e at a high potential). The main research direction for FAO catalysts is to suppress the dehydration path and to enhance the dehydrogenation path by modification of the physicochemical properties of Pt by alloying with other secondary metals like Au, Bi and Sb [17–23] or by attaching the secondary metals on Pt surfaces [24–31].
One of the intensively studied examples to enhance FAO performance is modification of Pt surfaces with Bi. For instance, an enhancement of Pt nanoparticles and Pt deposits on Au nanoparticles by factor of 5 has been demonstrated using a modification of the Pt surfaces with Bi using irreversible adsorption method [32]. On Bi-modified Pt surfaces, in particular, an additional FAO path (named “oxidized surface paths”) at a potential range overlapping with that of the dehydrogenation path was reported to take place as a third FAO path [33]. On the other hand, a power of 300 W was generated with a DFAFC stack of 35 membrane electrode assemblies, which was possible with a mass production of Bi-modified Pt nanoparticles using irreversible adsorption method [34]. Recently, deposition of additional Pt on Bi-modified Pt nanoparticle further improved FAO performance by factor of 3 as well as catalytic durability [33]. In this particular point of view, combination of Pt and Bi may be a breakthrough toward practical DFAFCs.
This work compares the FAO catalytic performances on the Pt nanoparticle and disk electrodes with sequentially deposited Bi and Pt. The Bi-modified Pt nanoparticle and disk electrodes were similar to each other in terms of the electrochemical behaviour in 0.1 M H2SO4 solution without and with formic acid. Upon additional Pt deposition on the Bimodified Pt surfaces, however, the electrochemical behaviour becomes significantly distinguishable, which will be discussed from the view point of the difference in Pt substrate shape.
2. Experimental
The Pt electrodes employed in this work were Pt nanoparticle (Pt NP), polycrystalline Pt disk (Pt(poly) disk) and Pt single crystal bead whose (111) facet was exclusively observed (Pt(111) disk). The electrodes of Pt(poly) disk and Pt(111) disk were used for electrochemical measurements and STM experiments, respectively.
Pt NP was a commercial Pt nanoparticle on Vulcan XC-72R (HiSPEC2000, Alfa Aesar). The loaded amount of Pt on the carbon support was ~10 wt%, and the average particle size was 2.08±0.05 nm.
Pt single crystal beads were produced by melting a Pt wire (0.5 mm diameter, 99.99%, Aldrich) in a hydrogen and oxygen flame to beads of ~2 mm in diameter. One of the (111) facets of a single crystal bead (i.e., Pt(111) disk) was positioned to a tungsten tip during scanning tunneling microscopy (STM) measurement. Pt(poly) disk electrodes were obtained by cutting the bead crystals in arbitral directions to hemispheres and polishing down to 0.25 μm with suspended diamond pastes.
Deposition of Bi and Pt was achieved using irreversible adsorption method [35]. The particular method consists of two steps in general. The first step is irreversible adsorption of precursor ions on a Pt electrode by contacting a solution of the intended precursor ion with the Pt electrode, followed by rinsing to remove the precursor solution from the electrode. The next step is electrochemical or chemical reduction of the adsorbed ionic layers on the Pt electrode. Due to the different shapes of the studied Pt electrodes, different procedures were utilized although the main idea was identical. In the case of Pt NP, 0.1 g of Pt NP was mixed with a precursor solution for 24 h; the supernatant precursor solution after settling down the catalyst powder for 30 min was removed; and the settled catalyst powder was rinsed with water several times. Then the rinsed catalyst powder was chemically reduced using 0.019 M NaBH4 (≥98.5%, Sigma Aldrich) solution. The Bi solution for Pt NP were prepared by dissolving Bi2O3 (99.999%, Sigma Aldrich) in 0.1 M H2SO4 (95–98%, Alfa Aesar). The concentration of Bi3+ and Pt2+ were 2.0 and 1.0 mM, respectively. On the other hand, the Pt(poly) disk and Pt(111) disk were dipped into a precursor solution for 1 min without potential control. After a brief rinse with water, the adsorbed ionic layers on a Pt disk were electrochemically reduced at 0.1 V in 0.10 M H2SO4 solution for 10 min. The Bi precursor solution for Pt disks was Bi2O3-saturated 0.1 M H2SO4 solution. After deposition of Bi, Pt was sequentially deposited using 1 mM K2PtCl4 (98%, Sigma Aldrich) in 0.1 M H2SO4 solution using the same method as above.
Electrochemical measurements were carried out in different ways depending on the shapes of the Pt electrodes. The working electrodes of Pt NP were dried slurries (20 μL) of catalysts spread on a glassy carbon electrode. Each slurry was a mixture of a catalyst powder (0.5 wt%), 5% nafion solution (1 wt%, Wako), and water. All experiments for Pt NP were carried out in a rotating disk electrode configuration with a rotation speed of 1000 rpm. On the other hand, the polished surfaces of Pt(poly) disks were exposed to electrolytes by maintaining a meniscus position during electrochemical measurements. The electrolytes were the solutions of 0.10 M H2SO4 solution for electrochemical behavior studies and 0.5 M formic acid solution (98%, Wako) + 0.1 M H2SO4 solution for FAO investigations. A three-electrode system was used with a home-made Ag/AgCl reference electrode in 1.0 M NaCl and a Pt counter electrode. All potentials are reported against the reference electrode. It should be emphasized that the scan rates for Pt NP and Pt(poly) disk were 10 and 50 mV/s, respectively, so that only qualitative comparison of the voltammograms of Pt NP and Pt(poly) disk is appropriate.
An X-ray photoelectron spectrometer (XPS, K-Alpha+ XPS spectrometer, Thermo Scientific) was employed to determine the atomic ratio of Bi and Pt. Photoelectrons were generated using a monochromatic Al Kα X-ray beam (1486.7 eV), and the photoelectron energies were measured using a multichannel hemispherical electron energy analyzer operated at a constant pass energy of 50 eV.
The structural analyses for Pt NP were performed using a transmission electron microscope (TEM, JEM-ARM200F, Jeol). For STM measurements for Pt(111) disk, a Nanoscope V (Veeco Inc.) was operated at the condition of 0.1 V sample potential, 0.01 V tip potential and 0.8 nA tunneling current in 0.10 M H2SO4 solution with a polyethylene-coated tungsten tip (0.25 mm diameter, Aldrich).
3. Results and Discussions
3.1 Characterization of Pt/Bi/Pt NP and Pt/Bi/Pt disk
Fig. 1 shows typical Pt and Bi 4f XPS spectra and TEM image of Pt/Bi/Pt NP [33]. A detailed analysis of the peak positions in Fig. 1(a) validates that the Pt in Pt/Bi/Pt NP was in a mixed state of metal and oxides, while the Bi was in an oxidized state due to air exposure. The Bi/Pt ratios measured using XPS spectra of Pt/Bi/Pt NP and Bi/Pt NP were 0.38±0.02 and 0.43±0.02, respectively, indicating that an additional Pt deposition took place on Bi/Pt NP via irreversible adsorption. In addition, the particle size of Pt/Bi/Pt NP in Fig. 1(b) was 2.7±0.1 nm, larger than those of Bi/Pt NP (2.4±0.1 nm) and Pt NP (2.08 ±0.05 nm) (whose TEM images are not shown). The size increments corresponding to the deposition of Bi on Pt NP and Pt on Bi/Pt NP, respectively, support that each of the Bi and Pt layers was roughly one atomic layer thick in average. The structure of Pt/Bi/Pt NP was suggested to be a bi-layer of sequentially stacked Bi and Pt, or a two atomic layer thick layer of Pt-Bi alloy on Pt NP.
Fig. 2 shows typical Pt and Bi 4f XPS spectra of Pt/Bi/Pt(poly) disk and STM images of Bi/Pt(111) disk and Pt/Bi/Pt(111) disk. In Fig. 2(a), the chemical states of Pt and Bi in Pt/Bi/Pt(poly) disk were similar to those on Pt/Bi/Pt NP. Upon Pt deposition on Bi/Pt(poly) disk, the Bi/Pt ratio decreased from 0.15±0.02 to 0.11±0.01 as well. Note that the Bi/Pt ratios of Pt/Bi/Pt(poly) disk are much smaller than that of Pt/Bi/Pt NP, probably due to the difference in the physical shapes of the Pt substrates. Fig. 2(b) and (c) are the STM images of Bi/Pt(111) disk and Pt/Bi/Pt(111) disk, respectively. Fig. 2(b) reveals that the Bi deposits were double-layered islands of ~4 nm in size (the inset of Fig. 2(b)), uniformly covering ~70% of the Pt surface [36]. Additional deposition of Pt over Bi/Pt(111) disk greatly modified the morphology as demonstrated in Fig. 2(c). Specifically, the protrusions covering ~98% of the Pt(111) surface were in the sizes of 5–20 nm (the inset of Fig. 2(c)), probably because each Pt deposit simultaneously covered a small number of the Bi deposits to be a large protrusion. In addition, the heights of the protrusions ranged from one to three in atomic height. The protrusions of two and three atomic layer height (covering ~70% of the Pt(111) disk) are certainly the deposits of Pt over the pre-existing Bi. Meanwhile, the protrusions of monatomic height (covering ~30% of the Pt(111) disk) are the Pt deposits on the empty Pt sites uncovered by the Bi deposits.
3.2 Electrochemical Behavior
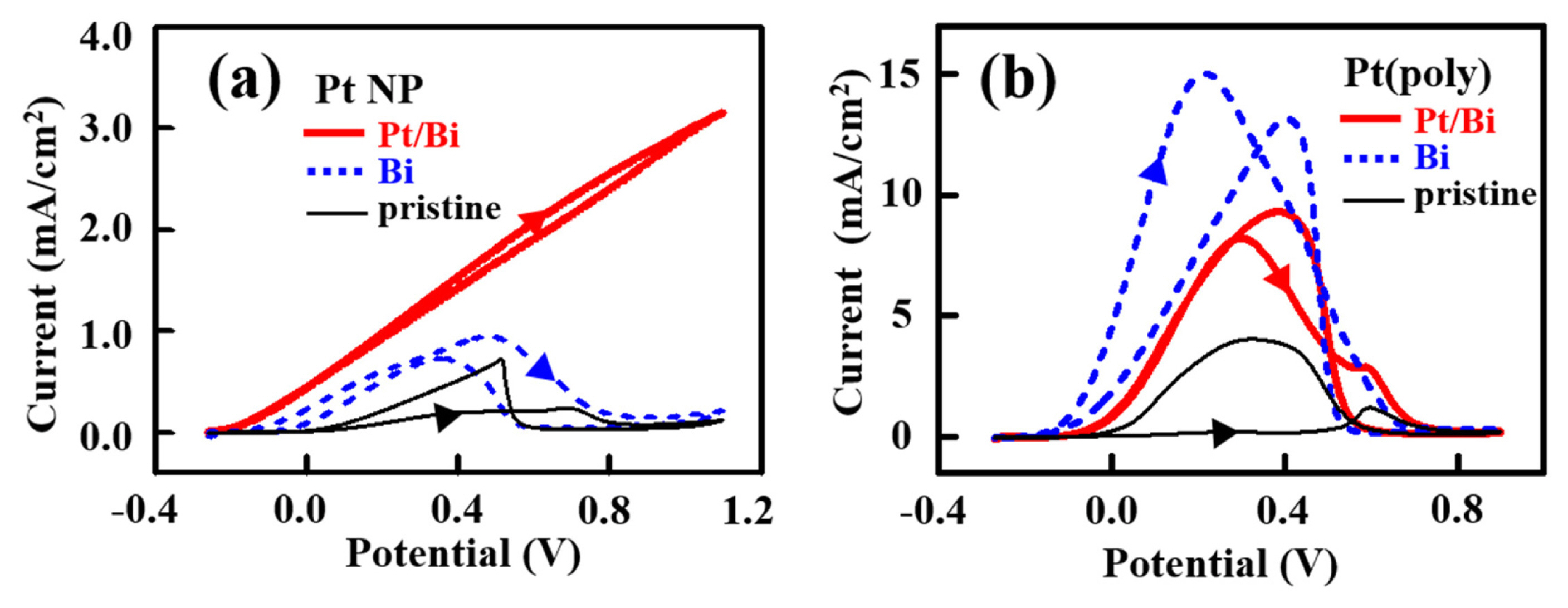
Fig. 3 compares the voltammetric behavior of pristine Pt, Bi/Pt and Pt/Bi/Pt. The thin solid line voltammograms of pristine Pt NP and Pt(poly) disk in Fig. 3(a) and (b) similarly showed electrochemical features of polycrystalline Pt, except for the large capacitive current coming from the carbon support of Pt NP. As demonstrated with the dashed line voltammograms in Fig. 3(a) and (b), the existence of Bi on both Pt NP and Pt(poly) disk reduced the charges of hydrogen, and an oxidation peak of Bi appeared at ~0.6 V. The Bi coverage, defined as the atomic number ratio of Bi to surface Pt atoms, was able to estimate using the hydrogen charges before and after Bi adsorption assuming that one Bi atom inhibits three Pt atoms from hydrogen adsorption [37]. The estimated Bi coverages on Bi/Pt NP and Bi/Pt(poly) disk were ~0.25. Additional deposition of Pt over Bi deposits altered the voltammograms of the Bi-modified Pt surfaces. The thick solid line voltammogram of Pt/Bi/Pt NP in Fig. 3(a) reveals that the hydrogen region did not increase, and that an additional surface oxidation current above 0.6 V (or after the Bi oxidation peak) appeared. The behavior of Pt/Bi/Pt(poly) disk was opposite to that of Pt/Bi/Pt NP as demonstrated with the thick solid line voltammogram in Fig. 3(b). Specifically, the hydrogen charge on Pt/Bi/Pt(poly) disk significantly increased closely to the value of pristine Pt(poly) disk. Furthermore, the Bi oxidation charge was suppressed seriously without the additional surface oxidation charge above 0.6 V.
The presented contrasting voltammetric behavior of Pt/Bi/Pt NP and Pt/Bi/Pt(poly) disk implies a difference in the interactions between Bi and additionally deposited Pt depending on Pt substrate. The microscopic images of Pt/Bi/Pt NP (Fig. 1(b)) and Pt/Bi/Pt(111) disk (Fig. 2(c)) do not provide a detailed distinction in structure enough to understand the contrasting electrochemical behaviors. In other words, the layers of Pt and Bi on Pt NP and Pt(111) disk may be identical. For instance, the layers of Pt and Bi on Pt NP can be either bi-layers stacked Bi and Pt in a layer-by-layer fashion or alloyed ones; the same logic is applicable to the layers of Pt and Bi on Pt(111) disk. Another example is the Pt deposits between the pre-existing Bi deposits, which are clearly demonstrated in Fig. 2(b) and (c). If the Bi deposits on Pt NP are islands of multiple layers as in Fig. 2(b), the morphology of Pt/Bi/Pt NP will be similar to that shown in Fig. 2(c). However, it is currently impossible with TEM to distinguish whether the Bi is homogenously spread over the surface of Pt NP or stacked up. In any case, the additionally deposited Pt is clearly intact with Bi, so that only the interaction(s) between the deposited Bi and Pt cannot provide a reasonable clue to understand the contrasting observations of Fig. 3. Then the unique difference between Pt/Bi/Pt NP and Pt/Bi/Pt disk is the shapes of Pt electrodes; i.e., nanoparticle of ~2 nm in diameter versus infinitely wide and thick disk. Because of the diminution of hydrogen adsorption and the enhancement of surface Pt oxidation, the interaction between Bi deposits and Pt overlayers is reasonably concluded to be stronger on Pt NP than on Pt(poly) disk. Or the interaction on Pt(poly) disk is so weak that the additionally deposited Pt expresses the electrochemical characteristics of Pt, not so much modified by the nearby Bi. Probably, the edges and vertexes of Pt nanoparticles or tensile stresses built up in the layers of deposited Bi and Pt on Pt NP are conceivable reasons for the strong interaction developed on Pt NP [38].
3.3 FAO behavior
Fig. 4 compares the FAO voltammetric behavior of pristine Pt, Bi/Pt and Pt/Bi/Pt. The FAO on Pt NP differs from that on Pt(poly) disk as shown with the thin solid line voltammograms in Fig. 4, although they were not modified. Specifically, in the positive-going scan there was a broad dehydrogenation peak at 0.4 V on Pt NP, which was absent on Pt(poly) disk. This difference implies that the dehydration path producing poisons was more sever on infinitely wide and thick Pt(poly) disk than on small discrete facets of Pt NP as demonstrated with the charge differences of the poison oxidation peaks at around 0.6 V [32]. In the following reverse scan, the currents via the dehydrogenation path on Pt NP and Pt(poly) disk initiated at 0.5 V. Deposition of Bi on the Pt electrodes modified the FAO behavior as presented with the dashed line voltammograms in Fig. 4. On both surfaces of Bi/Pt NP and Bi/Pt(poly) disk, the onset potential shifted from 0 V to −0.2 V, and the FAO current via the dehydrogenation path in the positive-going scan increased significantly. In addition, the positive ends of the FAO peaks in the positive-going scan extended to the potentials higher than 0.6 V (the peak potential of Bi oxidation in Fig. 3), indicating that the oxidized surface path became effective. In the following negative-going scan, the dehydrogenation path initiated at 0.5 V after the reduction of oxidized Bi-modified Pt surfaces. The FAO behavior of Bi/Pt NP and Bi/Pt(poly) disk suggests that Bi on Pt surfaces lowered the onset potential and suppressed the dehydration path. Also, the role of Bi regarding FAO was more effective on Pt(poly) disk judging from the FAO peak current ratios of the Pt electrodes without and with Bi [39]. The additional Pt deposition on the Bi-modified Pt surfaces changed the FAO behavior remarkably depending on the shape of Pt electrode as presented in Fig. 4. The thick solid line FAO voltammogram of Pt/Bi/Pt NP in Fig. 4(a) is practically a linear polarization curve with enhanced currents in the studied potential range. More specifically, the current below 0.6 V is due to the dehydrogenation path, and the current above 0.6 V is ascribable to the oxidized surface path. On the other hand, the thick solid line FAO current of Pt/Bi/Pt(poly) disk decreased as shown in Fig. 4(b). For instance, the peak current of the dehydrogenation path at ~0.3 V in the positive-going scan decreased roughly to a half of Bi/Pt(poly) disk, and the onset-potential shifted from −0.2 V to −0.1 V. In addition, the FAO current via the oxidized surface path above 0.6 V was not so significant as on Pt/Bi/Pt NP.
The FAO performances on the layers of Pt and Bi evidently depend on the shapes of Pt electrodes. The strong interaction between the deposited Pt and Bi on Pt NP is highly related to the FAO enhancement via the promotion of the dehydrogenation path below 0.6 V and the oxidized surface paths above 0.6 V, while the weak interaction on Pt(poly) disk is connected to the slight decrease in FAO performance. Here, it should be noted that Pt/Bi/Pt(poly) disk is certainly better than pristine Pt(poly) disk, indicating that the Bi deposits with additional Pt enhance FAO. However, a Pt substrate also contributes somehow to the interaction between the deposited Bi and Pt depending on its shape, which is consistent with the previous report from our group [32,39]. The same reasons discussed previously for the contrasting electrochemical behavior would explain the effect of Pt substrate shape on FAO in the presence of Bi. Thus this work clearly demonstrates that the shape of Pt substrate should be seriously considered in designing FAO catalysts of Pt and Bi.
4. Conclusions
This work demonstrated that the FAO catalytic performances of the Pt and Bi layers deposited on Pt substrates strongly depended on the shapes of the Pt substrates. The Bi deposits on Pt NP and Pt(poly) disk certainly enhanced FAO performance in a similar way. The additionally deposited Pt on the Bi deposits changed the electrochemical behavior and FAO activity in a different way depending on the shape of the Pt substrates. The Pt overlayers on Bi deposits on Pt NP significantly enhanced FAO performances, while those on Pt(poly) disk slightly decreased. These observations were understood in terms of different interactions between the Pt over-layers and Bi deposits greatly affected by the shapes of Pt substrates.