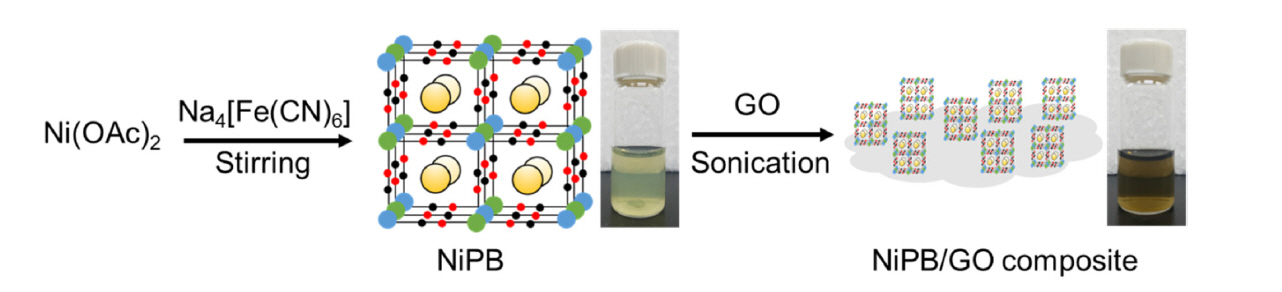
1. Introduction
Growing environmental concerns related to radionuclides derived from nuclear activities and accidents have spurred the development of efficient separation and removal technologies for radionuclides. Among the main fission products, Cs radioisotopes such as 137Cs (t1/2 ~ 30 y) and 135Cs (t1/2 ~ 2,000,000 y) have attracted much interest because biochemical confusion and substitution of vital alkaline metals like Na and K with Cs poses significant biohazards to terrestrial and aquatic organisms [1]. The Fukushima Daiichi nuclear power plant disaster released tons of radionuclide-contaminated wastewater into the sea. Subsequently, extensive efforts have been made to remove radionuclides [2], especially Cs radioisotopes, from the environment [3]. Several methods such as ion-exchange [4], sorption [5], and solvent extraction [6] have been developed for Cs separation. However, these methods would have drawbacks such as requirement of expensive materials, high operation costs, production of large amounts of secondary waste, and poor removal efficiency.
Transition metal hexacyanoferrates (MHCF) including Prussian Blue (PB) and its analogues have found a wide range of applications such as electrochromic films, batteries, capacitors, and electrochemical sensors owing to their attractive electrochromism, ionexchange, molecular magnetism, and electrocatalytic characteristics [7–9]. These inorganic polymers feature a cyanide-bridged metal open framework, and the concomitant cubic lattice spaces show excellent adsorption capability for radioactive Cs in wastewater [10,11]. Such adsorbents are usually prepared in the form of very fine precipitates; this may require an elaborate filtration or centrifugation process to separate the Cs-adsorbing powder from aqueous solutions. Thus, they are often combined with functional support materials for their practical uses such as column packing [12]. Furthermore, oxidation state modulation of the MHCF framework can be used to control Cs sorption. MHCF films can electrochemically achieve ion separation and adsorbent regeneration, making them promising for reducing the volume of secondary waste such as Cs-adsorbing MHCF materials [13]. Recently, Chen et al. demonstrated column separation of Cs using the electrochemically switched ion exchange capability of Cu hexacyanoferrate [13].
Graphene-based materials have found a variety of applications such as electronics [14], sensors [15,16], catalysis [17,18], energy-storage devices [19–21], and drug delivery [22] owing to their superior electronic capacity, mechanical properties, and chemical stability, and the high specific surface area of their sp2-hybridized carbon atom layer [23,24]. Graphene oxide (GO), chemically very accessible and proccessible in the graphene family [25], can be easily combined with functional materials, excelling the properties of the composites in various areas [26,27]. For example, MHCF/GO nanocomposites have been used as H2O2 reduction sensors with rapid electrocatalytic response [28] and Cs adsorbents with excellent removing efficiency [29]. This type of ion-exchangeable and electrochemically active MHCF/GO system can be a candidate material for the electrochemical removal of radioactive Cs from wastewater.
Herein, we coated an indium tin oxide (ITO) electrode by drop-casting a mixture solution of nickel hexacyanoferrate and graphene oxide (NiPB/GO). Spectroscopic, electron microscopic, and X-ray diffraction analyses were used to determine the compositional and morphological properties of the NiPB/ GO composite film. We examined the electrochemical properties of the NiPB/GO-modified electrode in various alkali metal electrolytes and demonstrated its enhanced Cs uptake capability with high ion selectivity during electrochemical treatment.
2. Experimental
All chemicals were used as purchased: Ni(OAc)2·4H2O, Na4[Fe(CN)6·10H2O], polyvinyl alcohol (PVA, Mw: 146,000–186,000), and LiNO3 from Sigma-Aldrich; KNO3 and CsNO3 from Alfa Aesar; NaNO3 from Hayashi Pure Chemical Ind., Japan; and GO powder (GO-V30, C: 45%–50%, O: 45%–50%) from Standard Graphene, Korea. Deionized H2O (18.2 MW cm) was obtained from a Mili-Q system. A PVA solution (0.01 wt%) was prepared by dissolving in H2O at 80°C with vigorous stirring for 2 h.
NiPB was prepared as follows. 2.7 g of Ni(OAc)2·4H2O was dissolved in a mixture of 175 mL of H2O and 25 mL DMF (Solution A). 4.8 g of Na4[Fe(CN)6·10H2O] and 7 g of NaCl were dissolved in 175 mL of H2O (Solution B). Solution A was added dropwise to Solution B with stirring at room temperature. The solid was separated by centrifugation and then washed with MeOH. The resulting greenish powder was dried overnight in an oven at 60°C. A NiPB/GO composite solution (0.5 mg/mL) was prepared by dispersing the mixture of NiPB and GO (w/w, 96:4) in an aqueous solution containing PVA (0.01 wt%,) via ultrasonication (power: 200 W) for 10 min (Fig. 1). ITO electrodes were cut into 1×4 cm2 pieces and cleaned using acetone and deionized water with utrasonication for 5 min. NiPB/GO-modified electrodes were prepared by drop-casting the greenish-black NiPB-GO solution (200 μL) on the ITO electrodes (area: 1×2 cm2). Drying in a convection oven (60°C for 30 min) afforded NiPB/GO film-modified electrodes. NiPB-modified ITO electrodes as a control were also prepared by the same method as described above. All electrochemical experiments were performed in a three-electrode system using a GAMRY potentiostat/galvanostat. Ag|AgCl and platinum mesh electrodes were used as reference and counter electrodes, respectively. The charge density was calculated by integrating the area of a CV curve. The morphology of NiPB and NiPB/ GO were observed using field-emission scanning electron microscopy (FESEM; JEOL JSM-7000F) and transmission electron microscopy (TEM; JEOL JEM-2100F). Powder X-ray diffraction (PXRD) patterns were obtained from a Bruker D8 Advance diffractometer. Raman spectra were measured by using an ANDOR Shamrock SR500i spectrometer.
3. Results and Discussion
Among all metal hexacyanoferrates, nickel hexacyanoferrate (NiPB) was chosen in this study because it shows reversible and reproducible electrochemical responses in alkali metal electrolytes [30,31] in addition to straightforward synthesis and high Cs adsorption capacity [12,32]. NiPB has an open framework with a face-centered crystal structure in which iron and nickel ions are linked by cyanide ligands. To achieve charge neutrality, the negatively charged NiPB framework allows guest cations such as alkali ions (Li+, Na+, K+, Ru+, and Cs+) to occupy the interstitial sites around the center of each cube, whereas the oxidation of Fe(II) to Fe(III) releases guest cations as described below:
where M is alkali metals such as Li, Na, K, Rb and Cs. Therefore, the oxidation state control of NiPB enables the uptake and release of counter cations (Fig. 2).
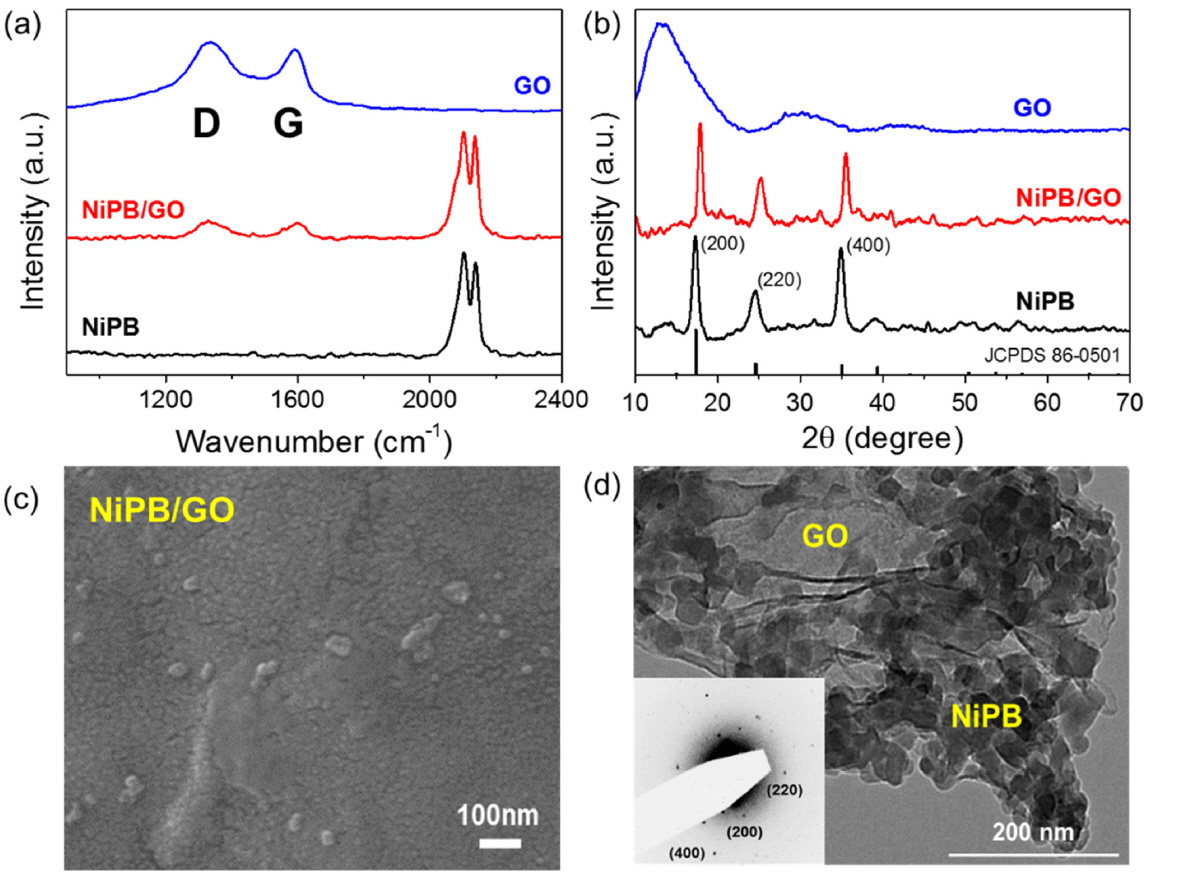
The NiPB/GO composite was characterized by Raman spectroscopy, PXRD, SEM, and TEM. Fig. 3a shows the Raman spectra of GO, NiPB, and NiPB/ GO. The GO shows two D and G bands at 1328 and 1590 cm−1 that represent the disordered carbon arising from structural defects and tangential C-C stretching vibrations, respectively [33,34]. NiPB shows two prominent bands peaked at 2137 and 2102 cm−1, corresponding to the stretching modes of the cyano bridging ligand coordinated to Fe(II) and Ni(II) [35], respectively. The Raman signals of the NiPB/GO composite appear at the same energy for the vibrations originating from both constituent materials, with NiPB bands having much higher intensity owing to the composition.
Fig. 3b shows XRD patterns of GO, NiPB, and NiPB/GO. A broad diffraction signal observed from 10° to 25° with GO is associated with the (002) plane. The NiPB shows diffraction peaks at 17.4°, 24.6°, and 34.9° that are respectively indexed to (200), (220), and (400) reflections of the face-centered cubic structure of the NiPB [32]. For the NiPB/GO composite, all characteristic peaks resemble the NiPB diffraction pattern. Although the signals associated with GO were not prominent, the (002) plane of the graphitic carbon seemed to overlap with the (220) plane of the NiPB nanoparticles.
The SEM image of NiPB/GO in Fig. 3c shows an assemblage of uniform NiPB nanoparticles, where GO is rarely observed owing to small amount of the GO (~4 wt%) in the composite. However, the representative TEM image in Fig. 3d clearly shows that cubic NiPB nanoparticles are decorated with GO. The inset of Fig. 3d shows the polycrystalline diffraction patterns indexed to the (200), (220), and (400) planes that are assigned to NiPB. Therefore, these characterization results indicate that the NiPB/GO composite can be formed via a simple preparation method.
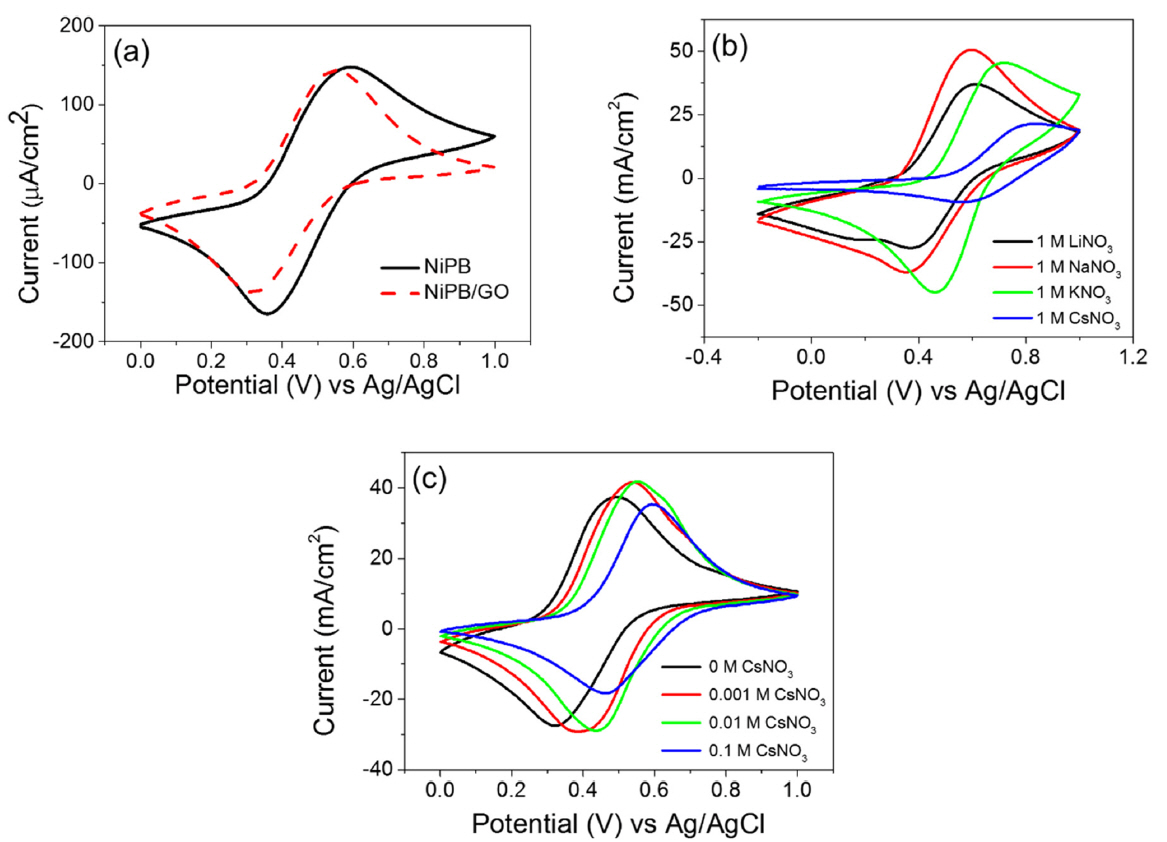
The electrochemical behavior of the NiPB/GO-modified electrode was examined in alkali metal electrolytes. Fig. 4a shows cyclic voltammograms (CVs) of the NiPB- and NiPB/GO-modified electrodes in 1 M NaNO3 electrolyte. The oxidation and reduction peaks of NiPB appear at 0.58 and 0.35 V vs. Ag|AgCl, respectively; these are attributed to the reversible redox couple of the iron center in the hexacyanoferrate complex coinciding with the deintercalation and intercalation of Na+ ion at the interstitial site of the NiPB framework (Eq. 1). The redox peaks of the NiPB/GO composite cathodically shift by 30–40 mV along with a slight peak current decrease, indicating that the addition of a small amount of GO slightly influences the redox properties of NiPB within the composite.
Fig. 4b shows CVs of the NiPB/GO-modified electrode in different alkali metal electrolytes. The redox couple of the NiPB appeared at 0.3–0.9 V vs. Ag|AgCl, the potential of which depended on the electrolyte used. E1/2 values for the NiPB/GO-modified electrode were in the order of Li ≈ Na < K < Cs (Table 1). This sequence qualitatively agrees with the results for MHCF-modified electrodes, and it can be explained by the interaction of the NiPB/GO film and the hydrated alkaline cations [36,37]. The highest redox potential in the CsNO3 electrolyte implies that the intercalation of Cs+ to the NiPB framework is thermodynamically more favorable than that of other alkali metals [37].
The electrochemical response of the NiPB/GO-modified electrode was also examined in mixtures of Na and Cs electrolytes. In Fig. 4c, the NiPB/GO-modified electrode shows that the current peak potential shifts to a positive value with increasing Cs+ concentrations in a series of electrolyte mixtures containing a different ratio of Na+ and Cs+, indicating that the composite film has good selectivity for Cs+ over Na+ even in the presence of excess Na (e.g., [Na+]/[Cs+] = 100). These results indicate that the NiPB/GO-modified electrode shows excellent affinity to Cs+ ions, which is similar to that of electrodeposited NiPB films [38,39].
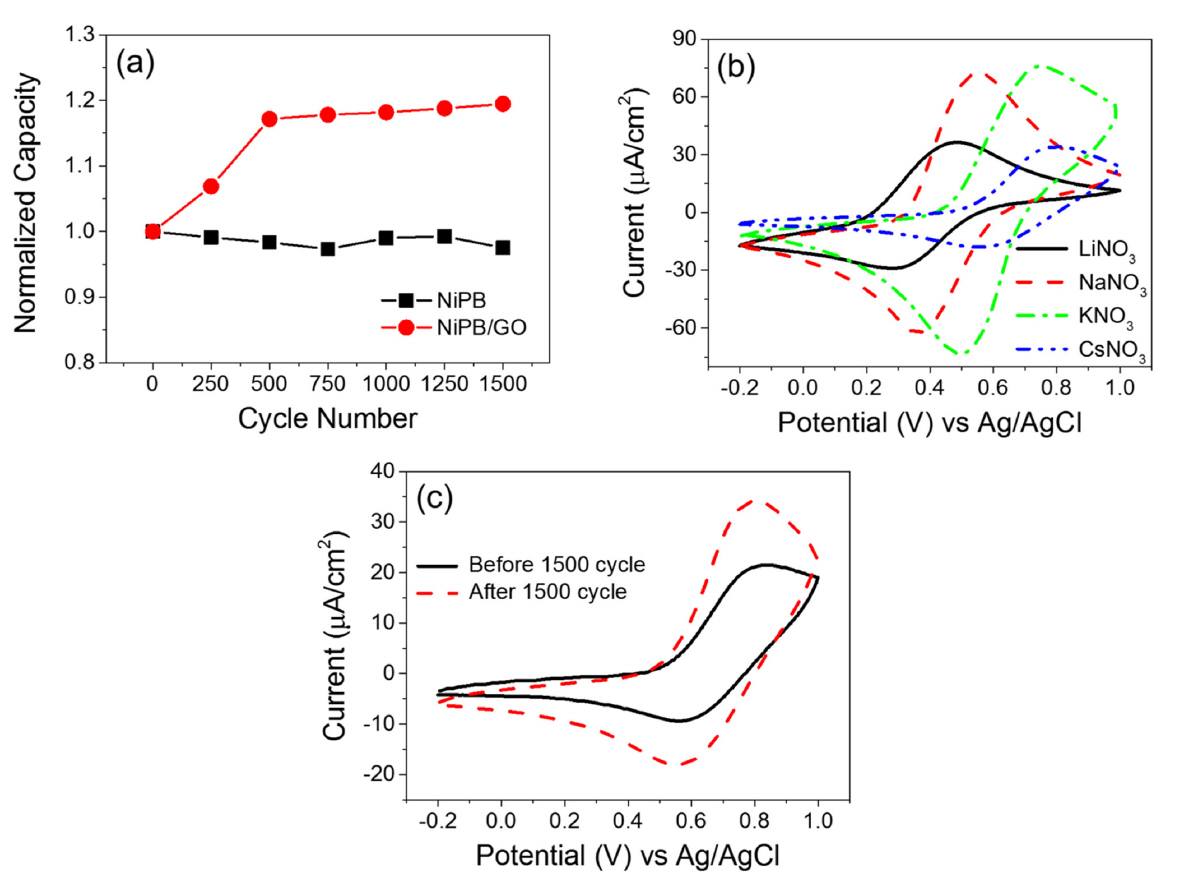
The regeneration performance of an electrochemical switched ion exchange system greatly depends on the stability of the electroactive film. The stability of NiPB- and NiPB/GO-modified films on ITO electrodes was evaluated by CV in a 1 M NaNO3 electrolyte solution in the potential range of 0–1 V at a scan rate of 100 mV·s−1 for 1500 cycles. The normalized capacity of the NiPB film prepared from the NiPB suspension in the PVA solution showed a slight decrease of ~2.5% after 1500 cycles (Fig 5a). This is somewhat different from the stability of NiPB films, which reportedly showed poor redox cycling stability owing to their gradual dissolution [40,41]. PVA seems to help NiPB particles stick to the electrode surface, thereby enhancing the long-term stability of the film during cycling.
Interestingly, redox cycling with the NiPB/GO-modified electrode improved the capacity of the composite film, unlike in the case of the NiPB-modified electrode. Fig. 5a shows that the normalized capacity of the NiPB/GO-modified electrode increases to 117% after the first 500 cycles and gradually increases up to ~120% after 1500 cycles in the NaNO3 solution in the potential range of 0–1 V at a scan rate of 100 mV·s−1.
If this type of electrochemically treated NiPB/GO (NiPB/GO-A) electrode can retain the ion selectivity of NiPB along with enhanced uptake capacity for Cs, it may serve as a promising solution for the electrochemical separation of radioactive Cs relative to the original composite. The electrochemical properties of the NiPB/GO-A electrode were examined in this context. Fig. 5b shows its CVs in different alkali metal nitrate electrolytes at a scan rate of 5 mV·s−1, and Table 1 summarizes the results. In the LiNO3 solution, the E1/2 value cathodically shifted from 0.49 to 0.38 V with nearly unchanged current density after electrochemical treatment. By contrast, in other alkali electrolytes, the E1/2 values marginally shifted (10–30 mV) and thus remained in the same order as in the originals after treatment, implying that the NiPB/GO-A electrode still showed ion selectivity for Cs over other alkali metals. Notably, the charge densities of the NiPB/GO-A composite apparently increased compared to those of the pre-treated electrode except in the case of the Li electrolyte. More interestingly, the increase in charge density augmented in the order of Na < K < Cs and the uptake capacity of the NiPB/ GO-A electrode for Cs was improved by nearly two times compared to that of the untreated one (Fig. 5c and Table 1).
Based on the behavior of the NiPB-modified electrode, this enhanced capacity of the NiPB/GO composite can be ascribed to the incorporation of GO into the NiPB film. The repetitive electrochemical ion insertion and desertion process through the NiPB film functionalized with the high-specific-surface-area GO likely improved guest ion accessibility in the composite electrode and may have softened the crystalline structure of NiPB/GO during the cycle [42,43]. Activation with large-sized hydrated Na ions could thus further enhance the accessibility and accommodation behavior of smaller hydrated ions such as K and Cs to the composite electrodes but not that of larger hydrated ions such as Li, resulting in the different extent of capacity enhancement with the cation species of the electrolytes in Fig. 5b.
4. Conclusions
We prepared a NiPB/GO-modified electrode by drop-casting a suspension of NiPB and GO (96:4 wt/ wt) onto an ITO electrode. Characterization of NiPB/ GO showed that NiPB nanoparticles were decorated with GO. Electrochemical studies of the NiPB/GO composite showed comparable electrochemically switched ion exchange capability and selectivity for alkali cations with respect to NiPB. Moreover, we found that the repeated potential cycling of the NiPB/ GO-modified electrode enhanced its cation uptake capacity. After 1500 cycles of electrochemical treatment in 1 M NaNO3, the Cs uptake ability of the NiPB/GO-modified electrode increased from 2.05 to 3.71 mC/cm2. In addition, the retained ion selectivity of NiPB for Cs may allow this electrochemically treated NiPB/GO composite to be a superior material for the electrochemical separation of radioactive Cs from wastewater over NiPB.